Genomic Annotation
Understanding the internals of annotation methodology


Research Article By
- Laya Babu (High School)
- Zaid Aafaq (High School)
Introduction
The information encoded into DNA is decoded via DNA sequencing. However, what happens after a genetic material is sequenced? The next step would be to annotate it, using genomic annotation. Genome annotation is the process of identifying the active parts of a genome, in other words, identifying all the parts of a DNA that code for something. This recent discovery has paved the way for many scientific breakthroughs and discoveries involving modern-day medicine. Genome annotation allows scientists to focus on a specific part of the DNA, rather than focusing on it as a whole. This allows for breakthroughs to be more quicker and efficient. Briefly, the process involves identifying the locations of genes, coding sections, and other functional elements to determine the significance of that sequence. After finding the genomic regions, biological information can be attached to each genome location. In the last few decades, genome annotation was used to identify long protein-coding genes of a single genome, but recently technology has advanced to accommodate the annotation of non-coding RNA genes, transposons, regulatory regions, pseudogenes, etc. According to Genome Annotation by Josep F. Abril and Sergi Castellano, “This increased resolution and inclusiveness of genome annotations (from genotypes to phenotypes) are leading to precise insights into the biology of species, populations and individuals alike.”
Genome annotation is classified into three levels
The nucleotide, protein, and process level. These topics will be briefly introduced. At the nucleotide level annotation, the main purpose is gene finding, as in finding the physical location of the DNA sequences. It allows for “the integration of genome sequence with other genetic and physical maps of the genome,” according to Genome annotation: from sequence to biology by Lincoln Stein. In nucleotide annotation, first, a sequence of a gene is found. Then it has to be named and then finally it is determined if it is a gene or a protein. The article mentioned also describes the function of protein-level annotation, which is “to assign function to the products of the genome,” and it determines the functions of genes. Also for the process-level annotation, “understanding the function of genes and their products in the context of cellular and organismal physiology is the goal.” So the last level identifies the pathways and processes with which the gene interacts.

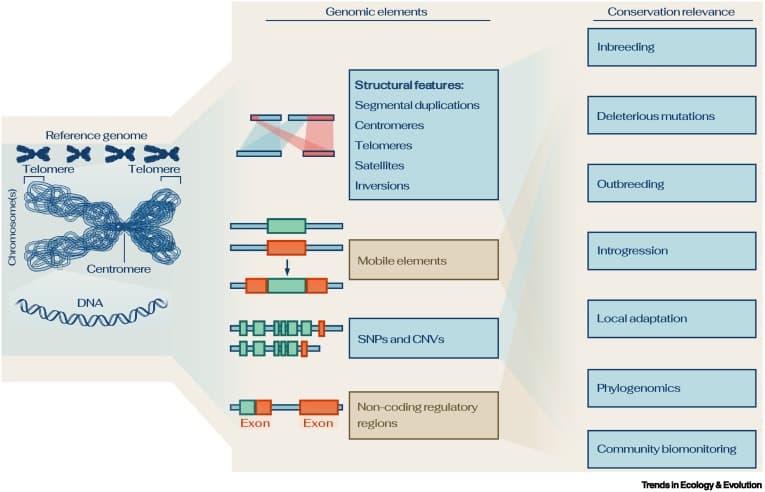
Reference genomes offer an (almost) complete record of the genome of a species. (source)
Genome annotation is divided into two steps
Structural annotation and functional annotation. Structural annotation refers to the identification of genomic elements like open-reading frames (ORFs) and their localization, gene structure, coding regions, and the location of regulatory motifs. On the other hand, functional annotations consist of attaching biological information to genomic elements, like biochemical function, biological function, involved regulation and interactions, and expression.
Programs used for Annotation
In structural annotation, Augustus is used for gene prediction and Aragon for tRNA and tmRNA predictions. While Augustus is used for eukaryotes and prokaryotes, glimmer3 can be used only for prokaryotes. In functional annotation, similarity searches can be done using BLAST, then the results can be used in other programs like Interproscan, WolfPSort, TMHMM, and BLASTGO for further analyses. Then antiSMASH is used for the identification of gene clusters.
Genome Annotations have become less accurate
According to Next-generation genome annotation: we still struggle to get it right by Steven L. Salzberg, genomic annotations are progressively getting less accurate as more draft genomes are produced. This is mainly because the automated annotations of the draft genomes are continuing to be difficult and errors in the draft assemblies affect the annotations as well. Especially for eukaryotes, gene finding is a seemingly difficult problem because genes are infrequently found between each other and there are several introns interrupting the way. So a partial solution would be RNA sequencing (RNA-seq). By aligning RNA-seq reads to a genome and then assembling those reads, we can construct a reasonably good approximation (including alternative isoforms) of the complete gene content of a species. However, this comes with numerous other problems, and they are summarized below:
- RNA-seq doesn’t accurately capture all genes as some may be expressed at low levels, expressed only in certain tissues, or be completely missed by the programs.
- Even if certain genes are expressed at high levels, there is no definite answer to whether they encode proteins or non-coding RNAs.
- Since draft genomes and their genes can be broken within many contigs, as the size of the contigs tends to be smaller than the size of the gene, it is still very difficult for the annotation software to know how to assemble the genes in order.

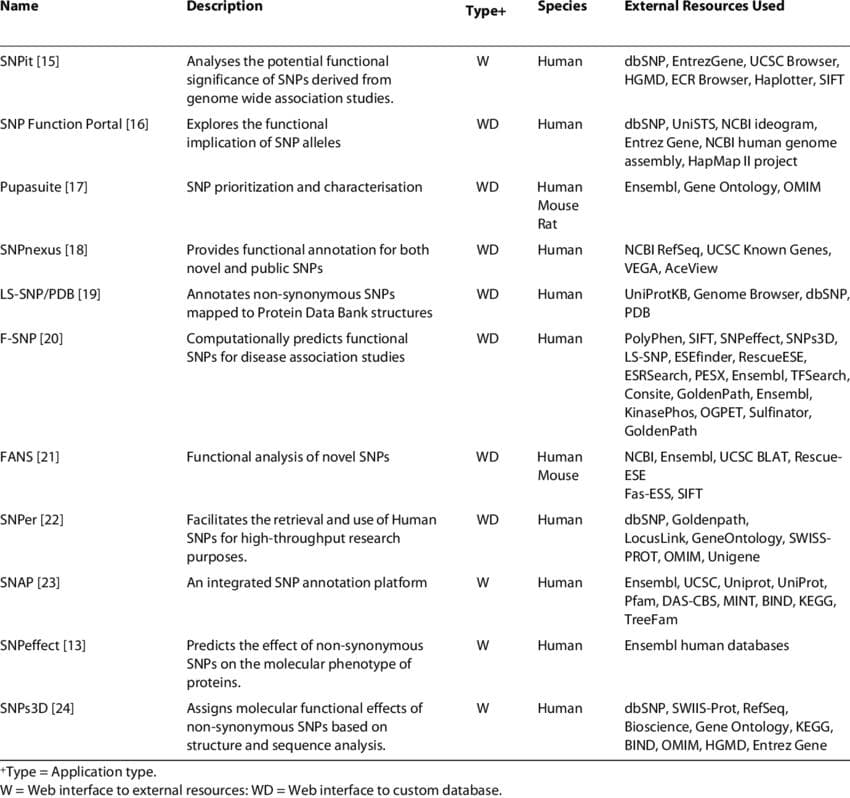
Existing SNP annotation tools. (source)
The next major problem is the actual contamination of the assembly, which can lead to errors in the annotation. Contamination refers to a few of the contigs that may not belong to be target species. “This presents one more (and growing) annotation challenge: when an annotation error is found and corrected in one species, any other annotation that relied upon it needs to be corrected as well.”
Functional Annotation
Through functional annotation, researchers can figure out what a portion of DNA is actually doing. This can't be stressed this enough, functional annotation opens the door to many, many, new and different opportunities in how we look at fighting disease and genetic disorders.
Because scientists now know exactly what a portion of a DNA is doing, they can accurately change it to combat a disease. This can also give rise to personalized treatments for a person. Functional annotation allows to understand the function of the gene and specific mutations in the specific genes. Most of the diseases have some link to DNA/genetics, and by identifying that connection you can develop treatments specifically for the person. Because every person doesn't have the same DNA, it doesn't make sense to try and create a cure for a genetic based disease that will suit all.

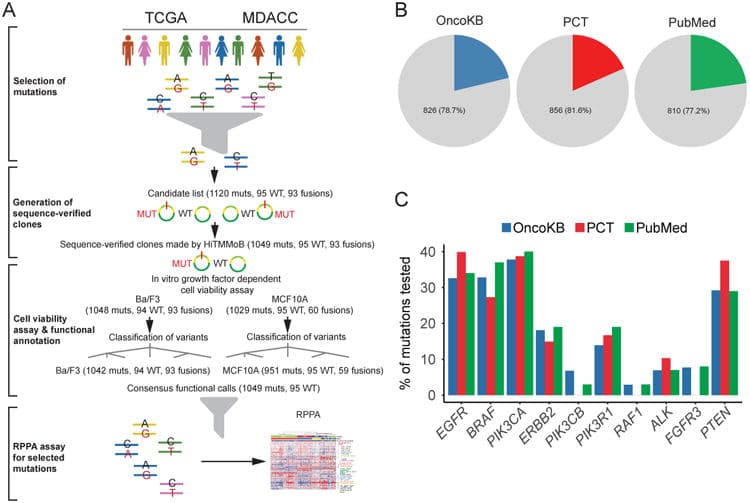
Overview of the functional genomic platform and cancer mutations tested (source)
(A) Mutations (muts), corresponding wild-type (WT) and fusion genes were selected from TCGA projects and MD Anderson Cancer Center patient databases. Clones were generated by the HiTMMoB approach, and tested in in vitro growth-factor dependent cell viability assays with Ba/F3 and MCF10A cell models. Mutations and wild-type variants were classified into functional categories based on these results. MCF10A cell lines stably expressing selected mutations were generated for reverse-phase protein array (RPPA) analysis. The numbers of mutant, wild-type and fusion constructs are annotated at each step. (B) Pie charts showing the proportions of the mutations annotated in OncoKB or Personalized Cancer Therapy (PCT) or PubMed literature among all the 1049 mutations tested. (C) Bar plots showing the literature coverage of mutations for the top 10 genes with the greatest number of mutations tested, as shown by the percentages of tested mutations per gene annotated in OncoKB or PCT or PubMed.
Functional Annotation in combating cancer
According to research done by the NCBI regarding functional annotation and cancer tumors they said, "Even for the most actively studied cancer genes such as PIK3CA, only a fraction of the variants identified in tumors have been functionally characterized." (NCBI, 2018 March 12, Systematic Functional Annotation of Somatic Mutations in Cancer - PMC) While we only fully understand a fraction of cancer tumors, this is still a great leap forward for cancer research because we are now starting to better understand cancer tumors and how to combat them. In order to understand the effect of various mutations found in various cancers, a tremendous amount of research is required. Essentially it entails an in-depth study of the genes and role of the stretches of DNA. Additionally, we need to understand how a gene interacts with another gene to affect the expression and disease.

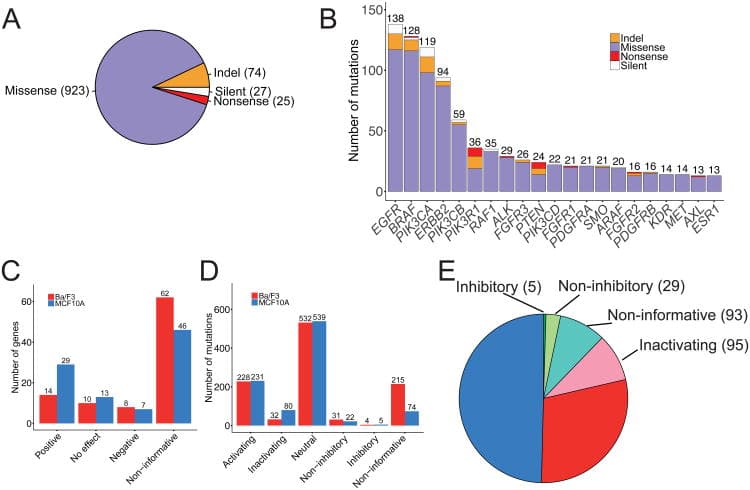
Functional annotation summary of wild-type genes and mutations (source)
(A) The numbers of missense (purple), indel (orange), nonsense (red) and silent (white) mutations tested are shown in parentheses. (B) The distribution of mutation types tested per gene for the 21 genes with >10 mutations tested is shown. (C, D) The functional annotations for wild-type genes (C) and mutations (D) in Ba/F3 (blue) and MCF10A (red) cell line models are presented based on the growth-factor independent cell viability assay results. (E) The number of mutations in each functional annotation is shown in parentheses. Eleven mutations with inconclusive functional annotations in Ba/F3 and MCF10A models were excluded.
Genome annotation for clinical genomic diagnostics
One of the most important discoveries of the 21st century, the complete sequencing of the human genome, has opened doors to a completely new pathway of how we approach diseases through genome annotation. This has allowed scientists to have a model reference normal human DNA sequence to compare other people's DNA. Now scientists have glimpse into where most of the introns and exons are located and what they do, making it easier to identify indels, SNP, and mutations in the DNA. This allows scientists to know what is present and what is missing in a person's DNA giving them a complete picture of a person's body systems and how they are functioning. This opens the road to so many more possible treatments for all types of genetic diseases because scientists can now identify what is potentially missing in a person's DNA that is causing a disease and work towards fixing it.
Article By:
-
Laya Babu (High School)
-
Zaid Aafaq (High School)
(External students, post on the request of Mentors)
References
- “DNA Annotation.” Wikipedia, Wikimedia Foundation, 26 Sept. 2022, https://en.wikipedia.org/wiki/.
- Erxleben, Anika, and Björn Grüning. “Galaxy Training: Genome Annotation.” Galaxy Training Network, Galaxy Training Network, 18 Oct. 2022, https://training.galaxyproject.org/training-material.
- Genome Annotation - North Dakota State University. https://www.ndsu.edu/pubweb/~.
- “Genome Annotation.” Genome Annotation - an Overview | ScienceDirect Topics, https://www.sciencedirect.com/.
- Libretexts. “7.13b: Annotating Genomes.” Biology LibreTexts, Libretexts, 3 Jan. 2021, https://bio.libretexts.org/.
- Salzberg, Steven L. “Next-Generation Genome Annotation: We Still Struggle to Get It Right - Genome Biology.” *BioMed Central*, BioMed Central, 16 May 2019, https://genomebiology..
- Stein, Lincoln. “Genome Annotation: From Sequence to Biology.” Nature News, Nature Publishing Group, https://www.nature.com/.
- Yandell, Mark, and Daniel Ence. “A Beginner's Guide to Eukaryotic Genome Annotation.” Nature News, Nature Publishing Group, 18 Apr. 2012, https://www.nature.com/.
- https://en.wikipedia.org/DNA_annotation
- https://en.wikipedia.org/wiki/Reference_genome
- https://www.cell.com/trends/
- https://www.sciencedirect.
- https://www.earlham.ac.uk/
- https://www.ncbi.nlm.nih.gov/
- https://www.sciencedirect.com/
- http://crdd.osdd.net/ga.php
The opinions expressed here are the views of the writer and do not necessarily reflect the views and opinions of Elio Academy.