Genetics of Epilepsy
By: Adina Almakhambetova, NIS PhM Almaty

What is epilepsy?
Epilepsy is a chronic noncommunicable brain condition that affects approximately 50 million people worldwide. Its main symptom is recurrent seizures, which can be brief bursts of involuntary movement impacting a specific part of the body (partial) or the entire body (generalized).
Seizure episodes occur due to excessive electrical discharges in clusters of brain cells. These discharges can originate from various brain areas. Seizures can range from minor muscular twitches or lapses in concentration to more severe convulsions lasting an extended period. The frequency of seizures varies, from infrequent occurrences (less than one per year) to frequent episodes (many per day).
A person is considered to have epilepsy if they meet any of the symptoms below:
- A diagnosis of epilepsy syndrome.
- At least two unprovoked or reflex seizures occurring >24 hours apart.
- Two unprovoked seizures happening during the course of the following ten years, followed by a chance of one unprovoked or reflex seizure and a likelihood of further seizures equal to the overall recurrence risk of at least 60%.
Diagnostic tests
To identify the source of the seizures and to diagnose epilepsy, patients undergo a number of tests. The assessment can consist of:
- A neurological examination. This examination looks at your behaviour, motor skills, mental health, and other areas to help diagnose and identify the type of epilepsy you could have.
- Blood examinations. Signs of infections, genetic disorders, or other illnesses that may be linked to seizures can be found in a blood sample.
- Genetic examination. Genetic testing may provide more information regarding epilepsy and treatment options for certain individuals with the illness. Although it is most frequently done on children, some adults with epilepsy may benefit from genetic testing as well.
Genetic tests will be explored further in the sections of each disease. You also may have one or more brain imaging tests and scans that detect brain changes:
- Electroencephalogram (EEG). The test that is most commonly used for epilepsy diagnosis. Electrodes are affixed to the patient's scalp during this test using a paste-like material or a cap. Their brain's electrical activity is then captured by the electrodes.
- It is usual for individuals to have abnormalities in the normal brain wave pattern if they have epilepsy. These alterations take place even in the absence of a seizure. Seizures can be recorded to assist identify the type of seizure the patient is experiencing or rule out other medical issues.
- High-density EEG. Patients may have a high-density EEG in a variant of the test, which places electrodes closer together than in a typical EEG. Your brain's damaged regions may be more accurately identified using a high-density EEG.
- MRIs, or magnetic resonance imaging. Strong magnets and radio waves are used in an MRI to provide a detailed image of the brain. An MRI examines the anatomy of the brain to identify potential seizure causes, just like a CT scan does . However, a CT scan offers a less detailed view of the brain than an MRI.
- Functional Magnetic Resonance Imaging (fMRI). Through the use of a functional MRI, doctors can quantify the variations in blood flow that take place during certain brain functions. Before surgery, this test can be used to pinpoint the precise locations of vital processes, such speech and movement, so that surgeons can avoid damaging those areas while performing their work.
- CT scan (computerised tomography). A CT scan creates cross-sectional pictures of your brain using X-rays. CT scans can identify brain cysts, tumours, or bleeding that may be the source of epilepsy.
- PET, or Positron Emission Tomography. PET scans visualise and identify changes in the brain's metabolic activity by injecting a small quantity of low-dose radioactive material into a vein. Low metabolism regions in the brain may be indicative of seizure hotspots. (https://www.mayoclinic.org/diseases-conditions/epilepsy/diagnosis-treatment/drc-20350098).
Treatment.
Treatment aims to reduce the frequency of seizure episodes or even eliminate them entirely for individuals diagnosed with epilepsy. Various treatment options are available:
- Medication: Anti-seizure medications, such as...
- Surgery: Epilepsy surgery involves removing the brain area responsible for generating seizures. It is particularly effective when seizures are localized in a specific region of the brain.
- Brain-stimulating therapies:
- Vagus Nerve Stimulator: This device is attached to the vagus nerve in the neck and implanted beneath the chest skin. It can reduce seizures by approximately 20-40%.
- Responsive Neurostimulation: Implantable devices analyze brain activity patterns to deliver medication or electrical charges.
- Deep Brain Stimulation: Electrodes implanted in targeted brain areas, usually the thalamus, are connected to a chest generator that sends electric signals to the brain.
- Ketogenic Diet: Some epilepsy patients have experienced symptom reduction by strictly following a high-fat, low-carbohydrate diet. This diet prompts the body to use fats for energy instead of carbohydrates. The exact mechanism behind its efficacy in reducing seizures is not fully understood; experts speculate that it may alter chemical processes in the body, leading to symptom suppression. (https://www.mayoclinic.org/diseases-conditions/epilepsy/diagnosis-treatment/drc-20350098)
Idiopathic vs Symptomatic Epilepsy.
Idiopathic (genetic) generalized epilepsy is a subtype of generalized epilepsy characterized by standalone generalised tonic-clonic seizures, which encompass both tonic (stiffening) and clonic (twitching or jerking) phases of muscle activity. It also includes specific conditions like childhood absence epilepsy, adolescent absence epilepsy, and juvenile myoclonic epilepsy.
On the other hand, symptomatic epilepsy emerges following a brain injury that carries the potential to trigger the condition. Examples of such injuries encompass severe head trauma, brain tumors, strokes, and more.
Historically, a wide array of epileptic disorders were categorized as symptomatic to differentiate them from idiopathic epilepsy, which is driven by hereditary factors. However, with the progression of genetic research, the clear demarcation between these two forms of epilepsy has become less distinct. Advances such as the identification of SCN1A mutations underlying severe myoclonic epilepsy of infancy (SMEI), a condition formerly categorized as symptomatic, have cast doubt on the strict classification between symptomatic and idiopathic cases. Additionally, the absence of major genes for common idiopathic epilepsies like juvenile myoclonic epilepsy challenges this classification.
Although hereditary epilepsies account for only a small proportion of seizure disorders, the etiology of most epilepsies is a complex interplay between acquired and inherited factors. This underscores the need for a more nuanced understanding of epilepsy classifications beyond the traditional idiopathic and symptomatic categories. (Andrade & Minassian, 2007).
Ion channel mutations in idiopathic epilepsy.
Idiopathic epilepsies, at least in the uncommon hereditary types, have been demonstrated to be mostly caused by malfunctions of mutant voltage- or ligand-gated ion channels. Ion-channel mutations are suspected of being a key factor in more prevalent epilepsies such juvenile myoclonic epilepsy and childhood and adolescent absence epilepsies, which are uncommon monogenic idiopathic epilepsies (Avanzini et al., 2007).
Mutations and Epilepsy
Autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE)
- The first gene for a hereditary type of idiopathic epilepsy to be identified (Schaffer et al., 1995).
- A significant Australian ADNFLE family that had previously contributed to the mapping of the condition to chromosome 20q13.3 was found to have a first mutation in the CHRNA4 gene in 1995 (Steinlein et al., 1995).
- Since then, further sleep-related frontal lobe epilepsy cases have been linked to mutations in two other genes (CHRNB2 and CHRNA2) as well as CHRNA4 itself.
- The 4- and 2-subunits of the neuronal nicotinic acetyleholine recepto r (nAChR) are encoded by CHRNA4, CHRNA2, and CHRNB2, respectively. The nAChRs are ligand-gated ion channel, distinguished by five homologous subunits (either same or different), which come together to form a cation-selective ion channel around a central axis.
- The second transmembrane domain contains the known ADNFLE mutations in CHRNA4 or CHRNB2. It appears that ADNFLE mutations preferentially target the channel's gating structure as the second transmembrane (and in some cases the third) domain primarily constructs the walls of the ion channel (Steinlein, 2007).
- The diagnosis of ADNFLE is established in a proband who has suggestive clinical findings and a family history consistent with autosomal dominant inheritance and/or a heterozygous pathogenic variant in CABP4, CHRNA4, CHRNA2, CHRNB2, CRH, DEPDC5, KCNT1, NPRL2, NPRL3, or STX1B identified by molecular genetic testing (https://www.ncbi.nlm.nih.gov/gtr/conditions/C1854335/).

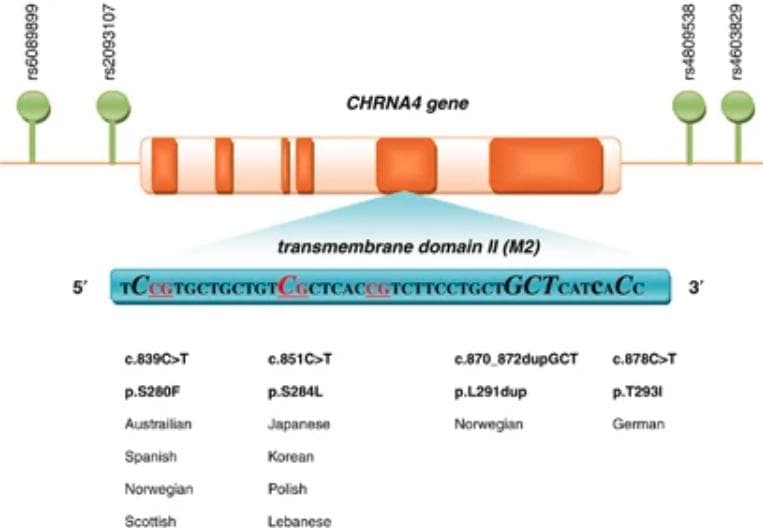
Summary of all the CHRNA4 mutations found in ADNFLE. The second transmembrane domain (M2) is where all of the mutations are found. CpG dinucleotide sites are underlined, and affected nucleotides are show n in bigger italic font.(Source)
Benign familial neonatal convulsions (BNFC)
- BFNC is a newborn seizure condition that is autosomal dominant.
- Several BFNC families have been reported to have mutations in the voltage-gated potassium channel genes KCNQ2 (chromosome 20q13.3) and (less often) KCNQ3 (chromosome 8q24) (Biervert et al., 1998; Charlier et al., 1998; Singh et al., 1998).
- Both genes develop channel subunits with the same six transmembrane regions (TM), voltage sensor in TM4, ion channel pore-building loop between TM5 and TM6, and lengthy C-terminal region containing sequence motifs for subunit assembly.
- For KCNQ2 and KCNQ3, respectively, more than 40 mutations have been documented thus far.
- The BFNC mutations are either truncating mutations (nonsense, insertion/deletions, or splice site mutations), massive deletions, or missense mutations in one of the TMs.
- Electroclinical events are suggestive of the disorder. In BFNE, neonates are neurologically normal and neurocognitive development is normal. Ictal electroencephalogram (EEG) may show focal interictal abnormalities, mainly over the central regions, but otherwise the EEG background is normal. The diagnosis is confirmed by genetic testing.
- The exact molecular genetic tests include targeted variant analysis, detection of homozygosity, sequence analysis of select exons, deletion/duplication analysis and sequence analysis of the entire coding region (https://www.ncbi.nlm.nih.gov/gtr/conditions/C3149074/).

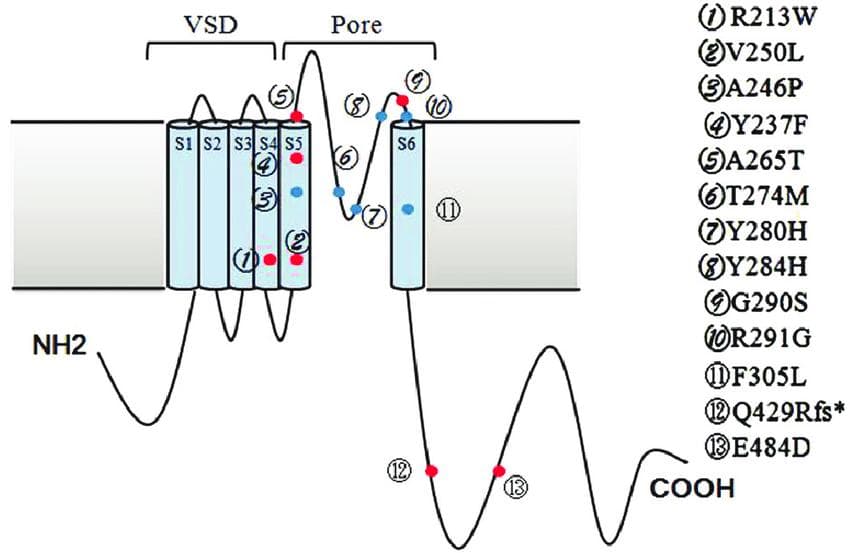
Structure and mutation locations of the channel protein KCNQ2 (K V 7.2). The structure of K V 7.2 is typical of voltage-gated potassium channel subunits; it consists of six transmembrane segments (S1–S6), an intracellular N- and C-termini, a voltage-sensing domain (VSD) composed by S1–S4, and a pore-loop between S5 and S6. The sites of KCNQ2 mutations in epilepsy that is easily managed are shown by red dots. The sites of KCNQ2 mutations in intractable epilepsy are shown by blue dots.(Source)
Benign familial infantile convulsions (BFIC)
- BFIC is an early childhood partial epilepsy condition that is inherited a utosomally dominantly (Vigevano et al., 1992).
- BFIC is a genetically heterogeneous condition that has been connected in several families to loci on chromosomes 1q23, 2q24, 19q, and 16p12-q12.
- Benign familial neonatal/infantile convulsions (BFNIC) are another uncommon seizure condition with an age of onset that falls between BFNC and BFIC. In BFNIC, the same family may experience seizures with neonatal and early infantile onsets.
- It has been established that changes in the voltage-gated sodium channel subunit gene SCN2A are the root cause of BFNIC, a gene that is also discussed as a minor gene for generalized epilepsy with febrile seizures plus (GEFS+) (Heron et al., 2002).
- There were also mutations in the proline-rich transmembrane protein 2 (PRRT2) gene located at 16p11.2 that can be associated with BFIC.
- Among the molecular genetic tests, sequence analysis of the entire coding region, deletion/duplication analysis, sequence analysis of select exons and targeted variant analysis can test this mutation (https://www.ncbi.nlm.nih.gov/gtr/conditions/C1853995/).

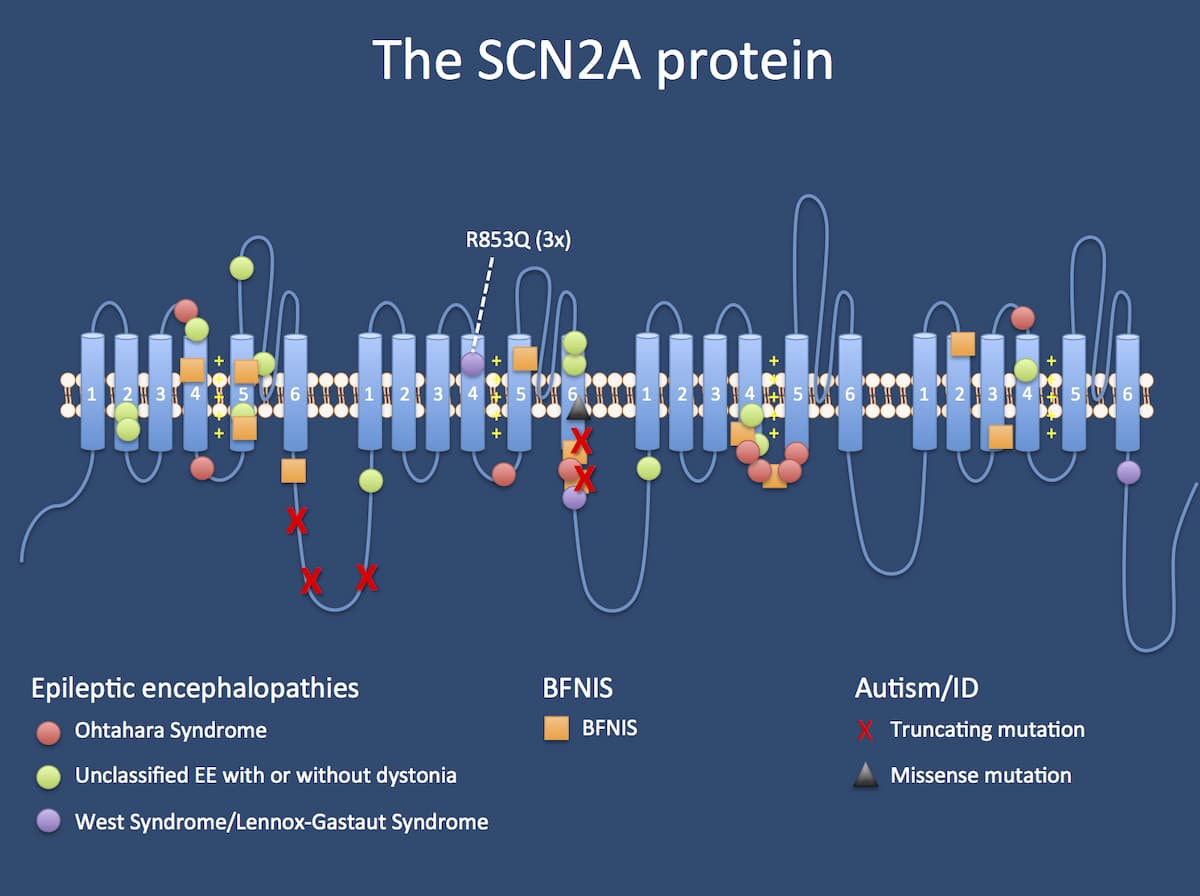
DNA vs phenotype. SCN2A-related phenotypes can be roughly divided into three groups: (1) mutations leading to Ohtahara Syndrome, unclassified epileptic encephalopathies with or without dystonia, Infantile Spasms, or Lennox-Gastaut Syndrome; (2) mutations leading to Benign Familial Neonatal-Infantile Seizures (BFNIS); and (3) mutations leading to autism and intellectual disability (ID).(Source)
Non-ion channel gene mutations in idiopathic epilepsy.
Numerous ion channels or proteins that influence ion-channel function are encoded by the majority of the epilepsy genes thus far listed (such as accessory channel subunits). The conclusion that idiopathic epilepsies are a category of channelopathies resulted from this. Non-ion channel genes, however, were shown to be at least marginally involved in the aetiology of idiopathic epilepsies.
Autosomal dominant lateral temporal lobe epilepsy (ADLTLE)
- Autosomal dominant partial epilepsy with auditory characteristics, often known as ADLTLE (syn. ADEAF), has been discovered to be caused by mutations in the LGI1 gene (leucine-rich glioma inactivated gene 1) (Kalachikov et al., 2002).
- On chromosome 10q24, the LG11 gene has a known mutational spectrum that includes both missense mutations and truncating variations.
- The LGI1 gene, which codes for a protein with seven so-called epilepsy-associated repeats (EARs) in the C-terminal half and a leucine-rich repeat motif (LRR) in the N-terminal end, is still mostly unknown in terms of its function. Most proteins that participate in a protein-protein interaction or a receptor function have the LRR motif (Staub et al., 2002; Gu et al, 2002).
- Clinical tests used: sequence analysis of select exons, sequence analysis of the entire coding region, deletion/duplication analysis and targeted variant analysis (https://www.ncbi.nlm.nih.gov/gtr/conditions/C4551957/)

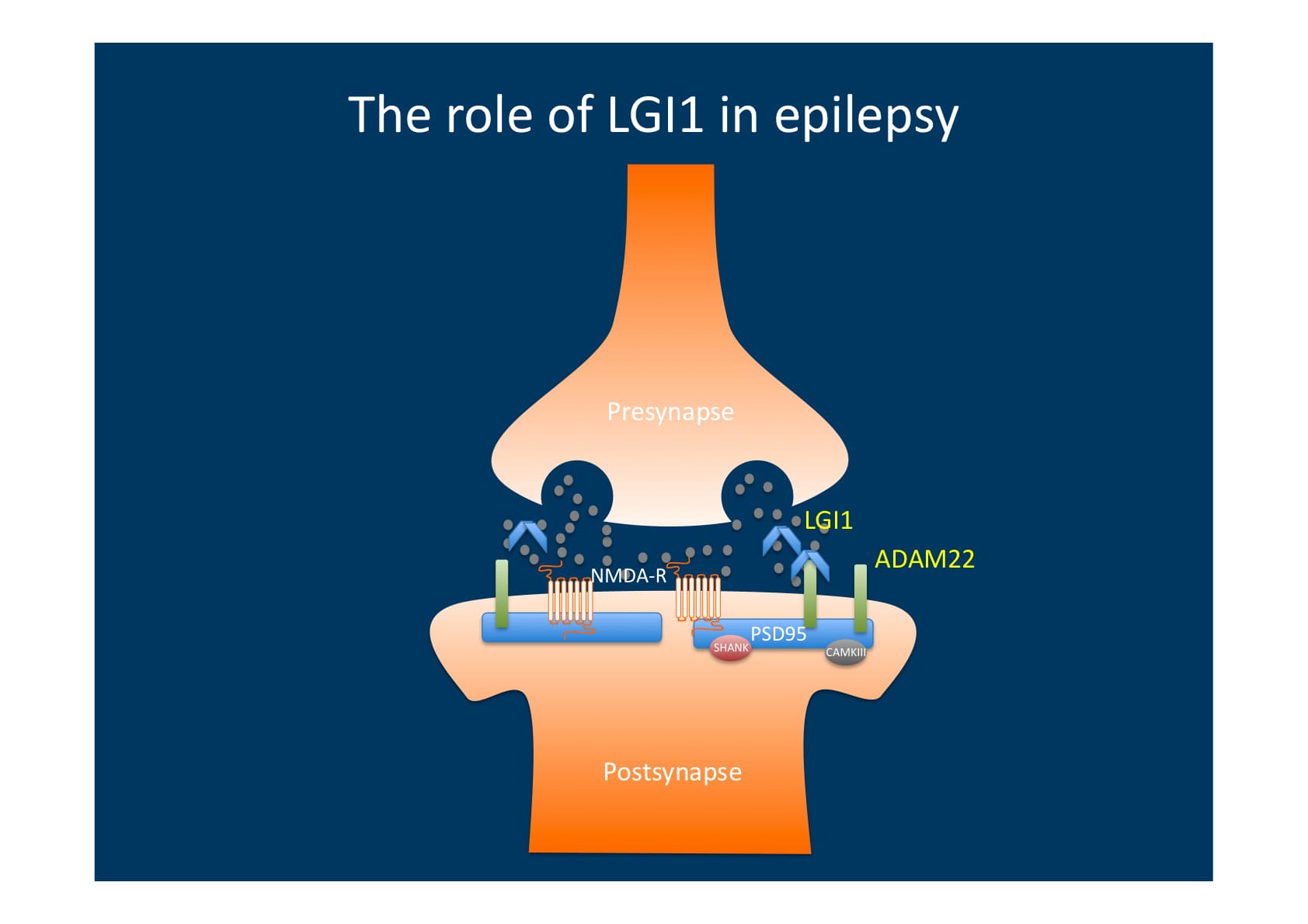
Postsynaptic ADAM22 is bound by LGI1, which is released from the presynapse. When ADAM22 is bound, the postsynapse experiences modified intracellular signalling that reduces excitability. This is most likely caused by subunit alterations in postsynaptic glutamate receptors. Hence, a net increase in excitability is caused by mutations in LGI that prevent ADAM22-mediated signalling.(Source)
Progressive myoclonus epilepsies (PMEs)
Myoclonus, a sudden, brief involuntary twitching or jerking of a muscle or group of muscles, generalised epilepsy, and progressive neurological deterioration, including dementia and ataxia (loss of muscle control and coordination), are characteristics of the uncommon clinically and genetically heterogeneous disorders known as PMEs (primarily autosomal recessive).
Unverricht-Lundborg disease (Baltic myoclonus), myoclonic epilepsy and ragged-red fibre disease (MERRF), neuronal ceroid lipofuscinosis (CLN), dentatorubropallidoluysian atrophy, etc. are examples of the diverse mechanisms that may underlie various neurogenetic syndromes characterised primarily by seizures and cognitive decline.
Unverricht-Lundborg disease (EPM1)
- The most prevalent kind of progressive myoclonus epilepsy is EPM1, sometimes referred to as Baltic or Mediterranean myoclonus epilepsy.
- It is an autosomal recessive neurological condition that typically manifests between the ages of 6 and 18 years.
- Various EPM1 patients have so far been shown to have a few point mutations in the CSTB gene (cystatin B, also known as stefin B), which is located on chromosome 21q22.3.
- However, in the majority of cases, a dodecamer repeat (a protein complex with 12 subunits) in the 5' flanking region of the CSTB gene has an unstable expansion that is responsible for the illness (Lafrenière et al., 1997).
- Can be diagnosed with targeted variant analysis, deletion/duplication analysis, sequence analysis of select exons, mutation scanning of select exons and sequence analysis of the entire coding region (https://www.ncbi.nlm.nih.gov/gtr/conditions/C0751785/).

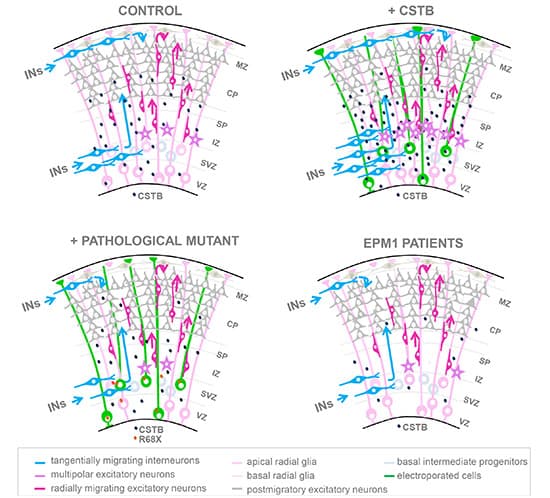
Patients with EPM1 epilepsy are affected by mutations in the cystatin B (CSTB) gene. In the mouse cortex and human cerebral organoids (hCOs), CSTB release stimulates the recruitment of migratory interneurons and fosters the expansion of progenitor cells. EPM1-derived hCOs have impairments in both roles. The overexpression of CSTB leads to the growth of progenitor cells in both the developing mouse brain and hCOs.
When migratory interneurons are recruited, CSTB is secreted. In the developing mouse cortex, there are fewer progenitors and migratory interneurons as a result of downregulating Cstb and overexpressing R68X. Cell non-autonomous reduction of proliferation occurs in cerebral organoids generated from EPM1. Cerebral organoids generated from EPM1 show early differentiation.(Source)
Future potential treatments for epilepsy.
- Subthreshold stimulation refers to the continuous stimulation of the seizure start zone. For certain individuals with seizures, subthreshold stimulation—continuous stimulation to a part of the brain below a level that is physically perceptible —appears to enhance seizure outcomes and quality of life. The use of subthreshold stimulation can prevent seizures before they start. Those with seizures that originate in the eloquent area of the brain —a region that cannot be removed due to its impact on speech and motor functions—may benefit from this therapy strategy. Alternatively, it may help those whose seizure characteristics make it unlikely that they would respond well to responsive neurostimulation therapy.
- TMS stands for transcranial magnetic stimulation. Without requiring surgery, TMS treats seizures by applying concentrated magnetic fields to specific brain regions. Patients who cannot benefit from surgery and whose seizures happen at the surface of the brain may utilise it.
- External trigeminal nerve stimulation. An apparatus that stimulates certain nerves to lessen seizure frequency works similarly to vagus nerve stimulation. In contrast to vagus nerve stimulation, however, the device is worn externally, negating the need for surgery to implant it. Studies have shown that external trigeminal nerve stimulation improves mood and seizure management.
- Less invasive surgical methods. Seizures may be treated using novel and minimally invasive surgical methods, such as MRI-guided focused ultrasound. Compared to open brain surgery, these operations have less risk for treating epilepsy (https://www.mayoclinic.org/diseases-conditions/epilepsy/diagnosis-treatment/drc-20350098).
Conclusion
Although the number of epilepsy genes that are now identified is outstanding; they likely just represent the very top of the iceberg. A rough estimate is that 50% of all genes are expressed in the brain at least during foetal development and may thus be considered seizure disorder possibilities. Aside from that, past studies suggest that variations in genomic DNA copy number and gene regulatory elements are likely to be just as significant for human illnesses as gene-related abnormalities (Ooi & Wood, 2007). Right now, whole-genome screening methods such as array-based comparative genomic hybridization (aCGH) or genome-wide single nucleotide polymorphism (SNP) analysis became important tools for the identification of genetic alterations with potential application to common forms of human epilepsy. The current generation of whole-genome screening techniques have emerged as crucial instruments for the detection of genetic changes that may have implications for the prevalent types of human epilepsy. It is also evident that, even with so much existing data and research about epilepsy, there's still a long way to go.

Click to view enlarged poster.

Click to read full paper.
Impact Statement

Hello, I’m Adina/Kai, from the mighty land of Kazakhstan! My project was a blend of both genetics and neurobiology respectively, carrying the name “Genetics of Epilepsy”. In it, I wanted to research the genetic basis for a widely researched, yet still greatly unknown neurological disease of epilepsy. With millions of people suffering from some form of an epileptic disorder, the etiology of this disease is especially important. Thanks to modern research, epilepsy has a lot of classifications regarding different aspects of epileptic symptoms. However, some epileptic disorders caused by genetic mutations are still a topic of further research. With Elio Academy’s help, I gained a new sense of community with people throughout the world. It motivated me to learn and represent a better world with my intentions. I hope that by sharing my project and what I’ve researched so far, I’ll help spark interest in groups of like-minded people who can change future medicine!
Student Reflection
References:
- Andrade, D. M., & Minassian, B. A. (2007). Genetics of epilepsies. Expert review of neurotherapeutics, 7(6), 727–734. https://doi.org/10.1586/14737175.7.6.727
- Avanzini, G., Franceschetti, S., & Mantegazza, M. (2007). Epileptogenic channelopathies: experimental models of human pathologies. Epilepsia, 48 Suppl 2, 51–64. https://doi.org/10.1111/j.1528-1167.2007.01067.x
- Scheffer, I. E., Bhatia, K. P., Lopes-Cendes, I., Fish, D. R., Marsden, C. D., Andermann, E., Andermann, F., Desbiens, R., Keene, D., & Cendes, F. (1995). Autosomal dominant nocturnal frontal lobe epilepsy. A distinctive clinical disorder. Brain : a journal of neurology, 118 ( Pt 1), 61–73. https://doi.org/10.1093/brain/118.1.61
- Steinlein, O. K., Mulley, J. C., Propping, P., Wallace, R. H., Phillips, H. A., Sutherland, G. R., Scheffer, I. E., & Berkovic, S. F. (1995). A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nature genetics, 11(2), 201–203. https://doi.org/10.1038/ng1095-201
- Steinlein O. K. (2007). Genetic disorders caused by mutated acetylcholine receptors. Life sciences, 80(24-25), 2186–2190. https://doi.org/10.1016/j.lfs.2007.03.007
- Biervert, C., Schroeder, B. C., Kubisch, C., Berkovic, S. F., Propping, P., Jentsch, T. J., & Steinlein, O. K. (1998). A potassium channel mutation in neonatal human epilepsy. Science (New York, N.Y.), 279(5349), 403–406. https://doi.org/10.1126/science.279.5349.403
- Charlier, C., Singh, N. A., Ryan, S. G., Lewis, T. B., Reus, B. E., Leach, R. J., & Leppert, M. (1998). A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nature genetics, 18(1), 53–55. https://doi.org/10.1038/ng0198-53
- Singh, N. A., Charlier, C., Stauffer, D., DuPont, B. R., Leach, R. J., Melis, R., Ronen, G. M., Bjerre, I., Quattlebaum, T., Murphy, J. V., McHarg, M. L., Gagnon, D., Rosales, T. O., Peiffer, A., Anderson, V. E., & Leppert, M. (1998). A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nature genetics, 18(1), 25–29. https://doi.org/10.1038/ng0198-25
- Vigevano, F., Fusco, L., Di Capua, M., Ricci, S., Sebastianelli, R., & Lucchini, P. (1992). Benign infantile familial convulsions. European journal of pediatrics, 151(8), 608–612. https://doi.org/10.1007/BF01957732
- Heron, S. E., Crossland, K. M., Andermann, E., Phillips, H. A., Hall, A. J., Bleasel, A., Shevell, M., Mercho, S., Seni, M. H., Guiot, M. C., Mulley, J. C., Berkovic, S. F., & Scheffer, I. E. (2002). Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet (London, England), 360(9336), 851–852. https://doi.org/10.1016/S0140-6736(02)09968-3
- Kalachikov, S., Evgrafov, O., Ross, B., Winawer, M., Barker-Cummings, C., Martinelli Boneschi, F., Choi, C., Morozov, P., Das, K., Teplitskaya, E., Yu, A., Cayanis, E., Penchaszadeh, G., Kottmann, A. H., Pedley, T. A., Hauser, W. A., Ottman, R., & Gilliam, T. C. (2002). Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nature genetics, 30(3), 335–341. https://doi.org/10.1038/ng832
- Staub, E., Pérez-Tur, J., Siebert, R., Nobile, C., Moschonas, N. K., Deloukas, P., & Hinzmann, B. (2002). The novel EPTP repeat defines a superfamily of proteins implicated in epileptic disorders. Trends in biochemical sciences, 27(9), 441–444. https://doi.org/10.1016/s0968-0004(02)02163-1
- Gu, W., Wevers, A., Schröder, H., Grzeschik, K. H., Derst, C., Brodtkorb, E., de Vos, R., & Steinlein, O. K. (2002). The LGI1 gene involved in lateral temporal lobe epilepsy belongs to a new subfamily of leucine-rich repeat proteins. FEBS letters, 519(1-3), 71–76. https://doi.org/10.1016/s0014-5793(02)02713-8
- Lafrenière, R. G., Rochefort, D. L., Chrétien, N., Rommens, J. M., Cochius, J. I., Kälviäinen, R., Nousiainen, U., Patry, G., Farrell, K., Söderfeldt, B., Federico, A., Hale, B. R., Cossio, O. H., Sørensen, T., Pouliot, M. A., Kmiec, T., Uldall, P., Janszky, J., Pranzatelli, M. R., Andermann, F., … Rouleau, G. A. (1997). Unstable insertion in the 5' flanking region of the cystatin B gene is the most common mutation in progressive myoclonus epilepsy type 1, EPM1. Nature genetics, 15(3), 298–302. https://doi.org/10.1038/ng0397-298
- Ooi, L., & Wood, I. C. (2007). Chromatin crosstalk in development and disease: lessons from REST. Nature reviews. Genetics, 8(7), 544–554. https://doi.org/10.1038/nrg2100
By: Adina Almakhambetova
The opinions expressed here are the views of the writer and do not necessarily reflect the views and opinions of Elio Academy.