Phenylketonuria (PKU)
Understanding the problems in brain development


By: Francesca Jimena Chevarría Gómez, Max Uhle Peruvian-German School
Introduction
Phenylketonuria (PKU) is an autosomal recessive genetic disorder, which means, two copies of the mutation must be present for the disease to develop. PKU can be caused by 400 different known variants in the PAH gene. The phenylalanine hydroxylase (PAH) enzyme performs the breakdown of the amino acid phenylalanine (phe) into tyrosine (Tyr), which is required by the body to produce stress neurotransmitters such as epinephrine, norepinephrine and dopamine. However, in the absence or deficiency of functional PAH, phenylalanine is not metabolized and accumulates to high levels in the blood, leading to problems in brain development.
PKU Condition
PKU or PAH deficiency is one of the most common and extensively researched monogenic Mendelian disorders in humans. The PAH gene is located on chromosome 12 and is responsible for the synthesis of the hepatic enzyme phenylalanine hydroxylase (PAH). When this mutation is present, the PAH enzyme is partially or completely ineffective, causing people with this disease to accumulate phenylalanine in all body tissues while lacking tyrosine. Since phenylalanine is not synthesized, it is degraded via an alternative metabolic pathway to phenylpyruvate, a neurotoxin that severely affects the brain during growth and development. Two types of PKU have been identified. The most severe is known as classical PKU, as described above. On the other hand, atypical or mild PKU is caused by a deficiency of tetrahydrobiopterin (BH4), a cofactor of the PAH enzyme.
Clinical symptoms
Phenylketonuria is typically identified shortly after birth. However, if left undiagnosed, patients will have high concentrations of phenylalanine in their blood and will excrete phenylpyruvic acid, leading to discolored urine. Due to this blood intoxication, neuronal activities are soon affected, and patients exhibit symptoms such as seizures, tremors, epilepsy, growth retardation, and hyperactivity. Other symptoms include halitosis and a foul odor from the skin or urine. Without treatment, neuronal damage is irreversible.

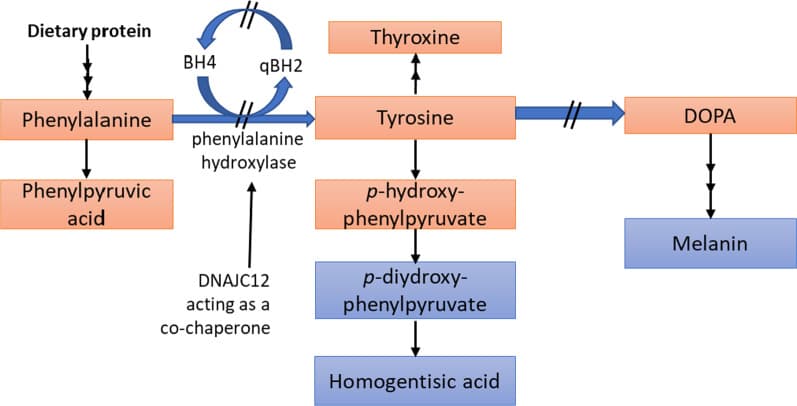
Phenylalanine metabolic pathways (Source)
Tests available
PKU is one of the diseases that has greatly benefited from newborn screening, enabling the assessment of Phe levels in newborns. The test involves pricking the heel and collecting blood. The collected sample is tested for various metabolic disorders, including PKU. If the initial test suggests that the baby may have PKU, further blood analysis is conducted to confirm the diagnosis.
Treatment available
Consuming protein-rich foods is hazardous for individuals with PKU because phenylalanine is abundant in these foods. A strict lifelong diet is the most common solution for PKU. The goal of the diet is to maintain blood Phe levels between 2 and 6 mg/dL (120 and 360 μmol/L). The phenylalanine-restricted diet can alleviate PKU symptoms and prevent brain damage. Additionally, this diet can be supplemented with glycomacropeptide (GMP), a protein component of whey that lacks phenylalanine and helps patients feel more satiated on the diet.

Phenylalanine is found in almost every food, except pure fat and sugar, which is a problem for PKU patients (Source)
Other treatment options include amino acid supplementation and therapies involving sapropterin and pegvaliase. For adults with PKU who cannot tolerate a restricted diet, amino acid supplements can be administered. These supplements compete with phenylalanine for transport in plasma, reducing its absorption and preventing it from crossing the blood-brain barrier to cause brain damage. Therapy with sapropterin is recommended for cases of atypical PKU. Sapropterin is a synthetic analogue of the cofactor BH4. This therapy activates the residual PAH enzyme, improving phenylalanine metabolism and reducing blood concentrations.
Finally, for adults with PKU who have uncontrolled blood Phe levels, they may opt for pegvaliase therapy. This is an enzyme replacement therapy that uses an enzyme pegvaliase. It converts phenylalanine to ammonia and trans-cinnamic acid, so that phenylalanine levels are reduced to normal ranges regardless of the activity of the PAH enzyme or the BH4 cofactor. Although effective, most patients experience side effects such as skin reactions, arthralgia and, rarely, anaphylactic responses.

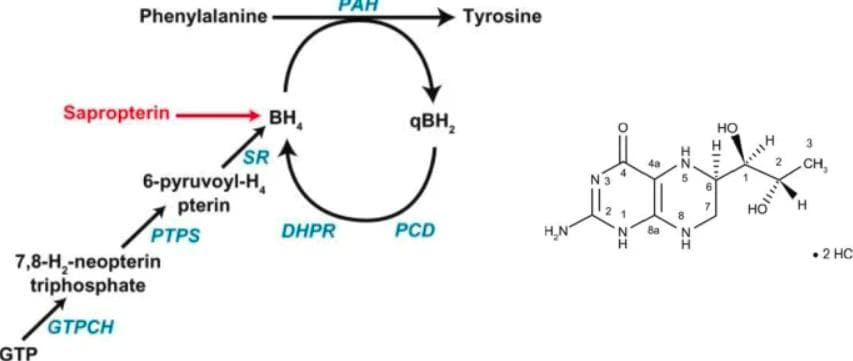
Functioning of sapropterin as a synthetic form of BH4 (Source)
Genetic basis of PKU
The PAH gene is situated on chromosome 12q23.1 and consists of 13 exons. Mutations in the PAH gene can vary in type and location, resulting in varying degrees of enzyme activity deficiency. Exon 12, 7, and 2 are the most commonly affected gene locations. While the gene is primarily expressed in the liver, it is also present in the kidneys and gallbladder.
Approximately 60% of cases of this disorder arise from a missense mutation, leading to the encoding of a different amino acid. This alteration changes the tertiary structure of the protein, rendering the active site unable to bind to the substrate and the PAH enzyme non-functional. In PKU, most patients are compound heterozygotes, carrying different mutations on each allele.
Gene therapy for PKU
Research is currently underway on new therapies for PKU, including gene therapy. Gene therapy aims to modify genes to treat diseases by repairing or replacing defective genetic material. This involves introducing a therapeutic gene into the patient's cells using viral or non-viral vectors.
In the case of PKU, the goal is to introduce a functional PAH gene into patients with bi-allelic pathogenic variants. Clinical trials are ongoing using viral vectors such as AAVHSC15 and AAV2/8. However, data from these trials are not yet available.

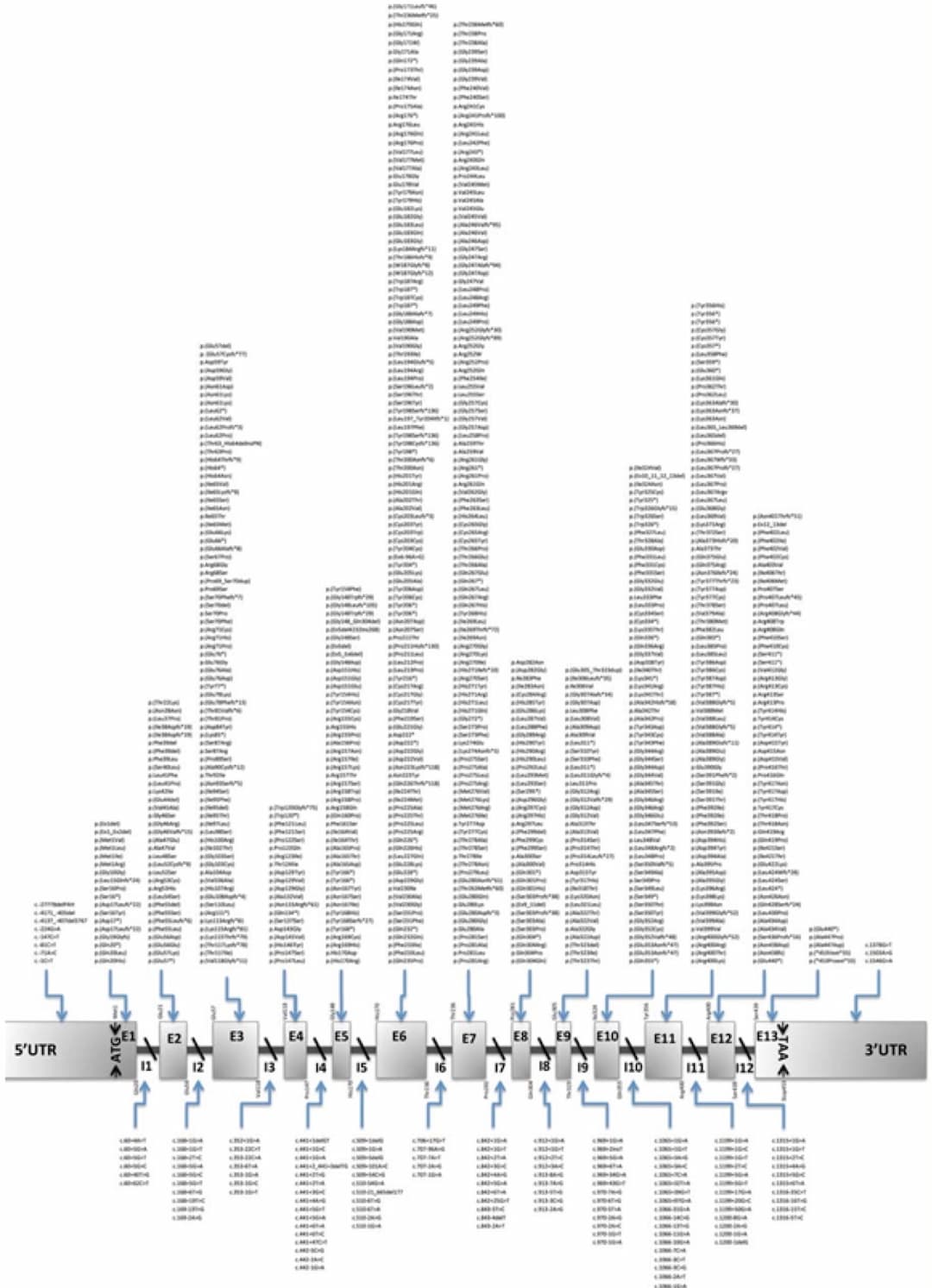
Physical structure of the phenylalanine hydroxylase gene with variants listed in the PAHvdb locus-specific database. (Source)
Lentiviral gene therapy is also being investigated as a potential option for treating PKU. Unlike viral vectors, lentiviral gene therapy would allow for the integration of the PAH gene into the host genome. Additionally, lentiviral vectors do not elicit pre-existing immunity, making all patients potential candidates for this therapy.
The genetic disorder PKU is one of the most extensively studied metabolic diseases. Recent advances have led to new treatments for PKU patients, and ongoing research is expected to continue. For instance, given that many neurological symptoms in PKU are linked to phenylalanine accumulation in the brain, research could focus on improving the delivery of enzymes or therapeutic agents across the blood-brain barrier.

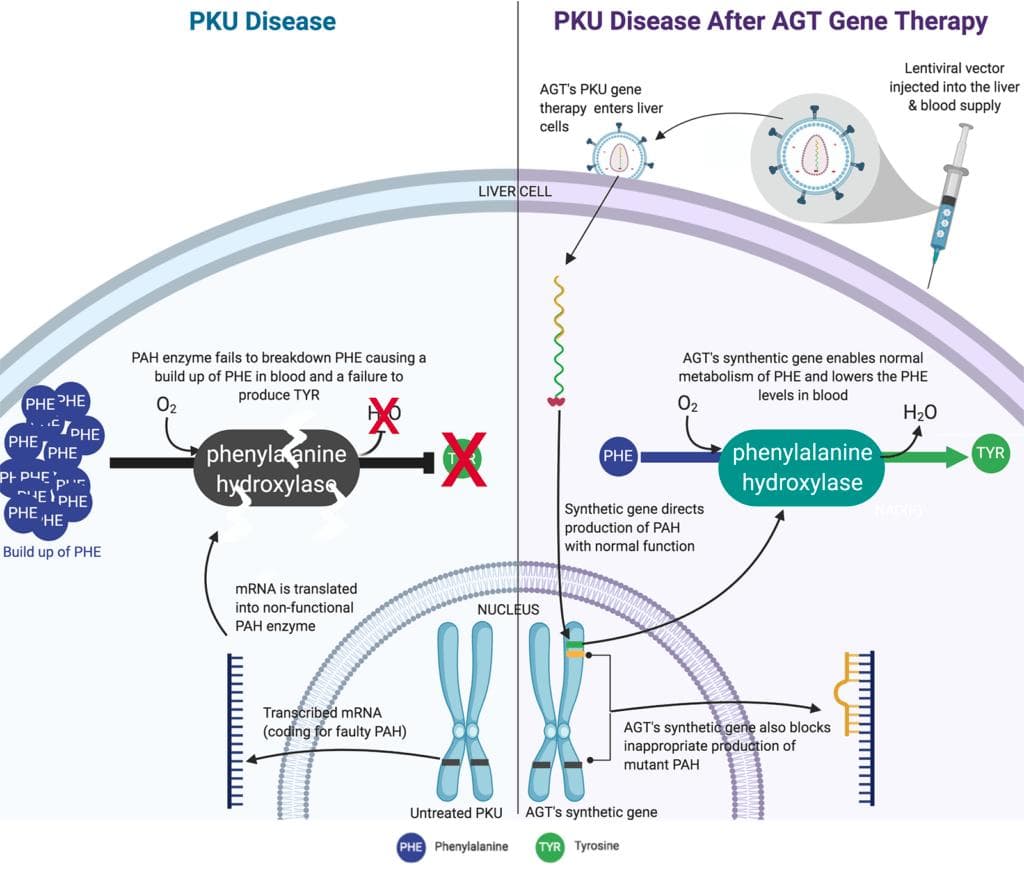
American Gene Technologies' Phenylketonuria Cure Program with Lentiviral gene therapy (Source)
Conclusion
The genetic disorder PKU is among the most extensively studied metabolic diseases. Recent decades have witnessed significant advances that have resulted in new treatments for PKU patients, and ongoing research is anticipated to persist. To illustrate, given that the majority of neurological symptoms in PKU are tied to the buildup of phenylalanine in the brain, research could be directed towards enhancing the transportation of enzymes or therapeutic agents across the blood-brain barrier.

Impact Statement

My name is Francesca Jimena Chevarría Gómez. I am a 2nd year IB student in Arequipa, Peru. I have always liked natural sciences and 3 years ago I decided that I wanted to study medicine. I entered Elio academy to deepen my knowledge in biomedical sciences and find out if this field really interested me. My experience during the two weeks of classes was fantastic and quite enriching. Despite the fact that many topics were difficult to understand, little by little with the help of the teacher I was able to understand them. To end the classes I made a report on a genetic disorder that had been interesting to me for a few months, phenylketonuria (PKU). The report talks about the disease, its symptoms, tests, treatments and new discoveries, as well as using the knowledge learned in the program to emphasize the genetic basis of the disease.
Student Reflection
References
Blau, N., Hennermann, J. B., Langenbeck, U., & Lichter-Konecki, U. (2011). Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Molecular genetics and metabolism, 104, S2-S9.
Blau, N., Shen, N., & Carducci, C. (2014). Molecular genetics and diagnosis of phenylketonuria: state of the art. Expert review of molecular diagnostics, 14(6), 655-671.
Blau, N. (2013). Sapropterin dihydrochloride for the treatment of hyperphenylalaninemias. Expert opinion on drug metabolism & toxicology, 9(9), 1207-1218.
Blau, N., Van Spronsen, F. J., & Levy, H. L. (2010). Phenylketonuria. The Lancet, 376(9750), 1417-1427.
Elhawary, N. A., AlJahdali, I. A., Abumansour, I. S., Elhawary, E. N., Gaboon, N., Dandini, M., ... & Kensara, O. A. (2022). Genetic etiology and clinical challenges of phenylketonuria. Human genomics, 16(1), 1-17.
Hillert, A., Anikster, Y., Belanger-Quintana, A., Burlina, A., Burton, B. K., Carducci, C., ... & Blau, N. (2020). The genetic landscape and epidemiology of phenylketonuria. The American Journal of Human Genetics, 107(2), 234-250.
Scriver, C. R., & Waters, P. J. (1999). Monogenic traits are not simple: lessons from phenylketonuria. Trends in genetics, 15(7), 267-272.
Thomas, J., Levy, H., Amato, S., Vockley, J., Zori, R., Dimmock, D., ... & PRISM investigators. (2018). Pegvaliase for the treatment of phenylketonuria: results of a long-term phase 3 clinical trial program (PRISM). Molecular genetics and metabolism, 124(1), 27-38.
Van Calcar, S. C., & Ney, D. M. (2012). Food products made with glycomacropeptide, a low-phenylalanine whey protein, provide a new alternative to amino acid–based medical foods for nutrition management of phenylketonuria. Journal of the Academy of Nutrition and Dietetics, 112(8), 1201-1210.
van Spronsen, F. J., Blau, N., Harding, C., Burlina, A., Longo, N., & Bosch, A. M. (2021). Phenylketonuria. Nature reviews Disease primers, 7(1), 36.
van Spronsen, F. J., & Enns, G. M. (2010). Future treatment strategies in phenylketonuria. Molecular Genetics and Metabolism, 99, S90-S95.
Van Vliet, D., van Wegberg, A. M., Ahring, K., Bik-Multanowski, M., Blau, N., Bulut, F. D., ... & van Spronsen, F. J. (2018). Can untreated PKU patients escape from intellectual disability? A systematic review. Orphanet journal of rare diseases, 13(1), 1-6.
Vockley, J., Andersson, H. C., Antshel, K. M., Braverman, N. E., Burton, B. K., Frazier, D. M., ... & Berry, S. A. (2014). Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genetics in medicine, 16(2), 188-200.
Yano, S., Moseley, K., & Azen, C. (2013). Large neutral amino acid supplementation increases melatonin synthesis in phenylketonuria: a new biomarker. The Journal of pediatrics, 162(5), 999-1003.
Project done at Elio Academy of Biomedical Sciences
By: Francesca Jimena Chevarría Gómez
The opinions expressed here are the views of the writer and do not necessarily reflect the views and opinions of Elio Academy.