Parkinson’s Disease
Understanding One of the Most Common Neurodegenerative Disorders


Background
Parkinson's disease (PD) is a progressive disorder of the nervous system that affects various regions of the brain, notably the substantia nigra, a region responsible for controlling balance and movement. The development of this disease is a result of a complex interplay of environmental and genetic factors, possibly involving multiple components that contribute to its onset. For instance, research has indicated that vascular insults to the brain, repeated head trauma, the use of neuroleptic drugs, exposure to pesticides, and manganese toxicity can elevate the risk of developing PD symptoms. Moreover, advancing age can introduce numerous stressors within the substantia nigra, thereby weakening neurons and their capacity to respond to further insults.
After Alzheimer's disease, PD stands as the second most common neurodegenerative disorder in the United States. While most individuals with Parkinson's develop the condition after the age of 60, around 5% to 10% experience its onset prior to the age of 50. Early-onset forms of Parkinson's are typically hereditary, and specific genetic changes have been associated with certain variants. Nonetheless, due to the heightened susceptibility to this condition with age and the increasing lifespan of the population, the number of PD cases in the United States is projected to double by the year 2040.
Numerous PD symptoms emerge when neurons in the substantia nigra, which typically generate dopamine, deteriorate or become impaired. Dopamine functions as a neurotransmitter, facilitating the transmission of signals within the brain to enable smooth physical movements. With the damage or loss of these neurons, communication between the brain and muscles becomes compromised, leading to reduced control over muscle movement.

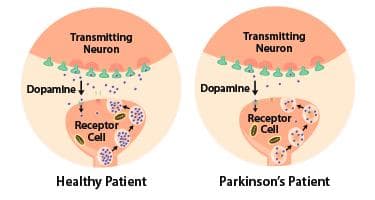
The diagram compares dopamine being transmitted to a receptor cell in a healthy patient and a patient with Parkinson's Disease. ([Source](https://www.ohsu.edu/brain-institute/understanding-parkinsons-disease))
Symptoms
Parkinson's disease gives rise to a multitude of symptoms, with the most prominent being involuntary or uncontrollable movements like tremors, stiffness, impaired balance, and coordination that can lead to falls. These symptoms generally begin gradually but worsen over time. Variability exists among different individuals in terms of symptom manifestation and the rate of progression. As PD advances, tasks like walking and speaking might become more challenging. Mental and behavioral changes, sleep disturbances, difficulties in eating, urinary and skin issues, depression, memory problems, and fatigue can also surface.
Diagnosis
Currently, no blood or laboratory tests exist for diagnosing non-genetic instances of Parkinson's disease. Medical professionals typically diagnose PD by examining an individual's medical history and conducting a neurological assessment. Improvement in symptoms following the initiation of medication can also serve as an additional indicator of the disease.
Treatment
While a cure for Parkinson's disease remains elusive, various treatments, including medications, surgical interventions, and other therapies, can alleviate certain symptoms. Medications help manage symptoms by augmenting dopamine levels in the brain, influencing other brain chemicals such as neurotransmitters that transmit information between brain cells, and addressing non-movement-related symptoms. The primary treatment for Parkinson's is levodopa, a precursor used by nerve cells to produce dopamine and replenish the diminishing supply in the brain. Typically, levodopa is taken alongside another medication called carbidopa, which mitigates or lessens some of the side effects of levodopa therapy (e.g., nausea, vomiting, low blood pressure, restlessness, etc.) and reduces the required dosage to improve symptoms. Additional treatments encompass dopamine agonists that stimulate dopamine production in the brain, as well as enzyme inhibitors like MAO-B inhibitors and COMT inhibitors that increase dopamine levels by slowing down the breakdown of dopamine in the brain. Ongoing research is also exploring other treatment avenues.
Genetics of Parkinson's disease
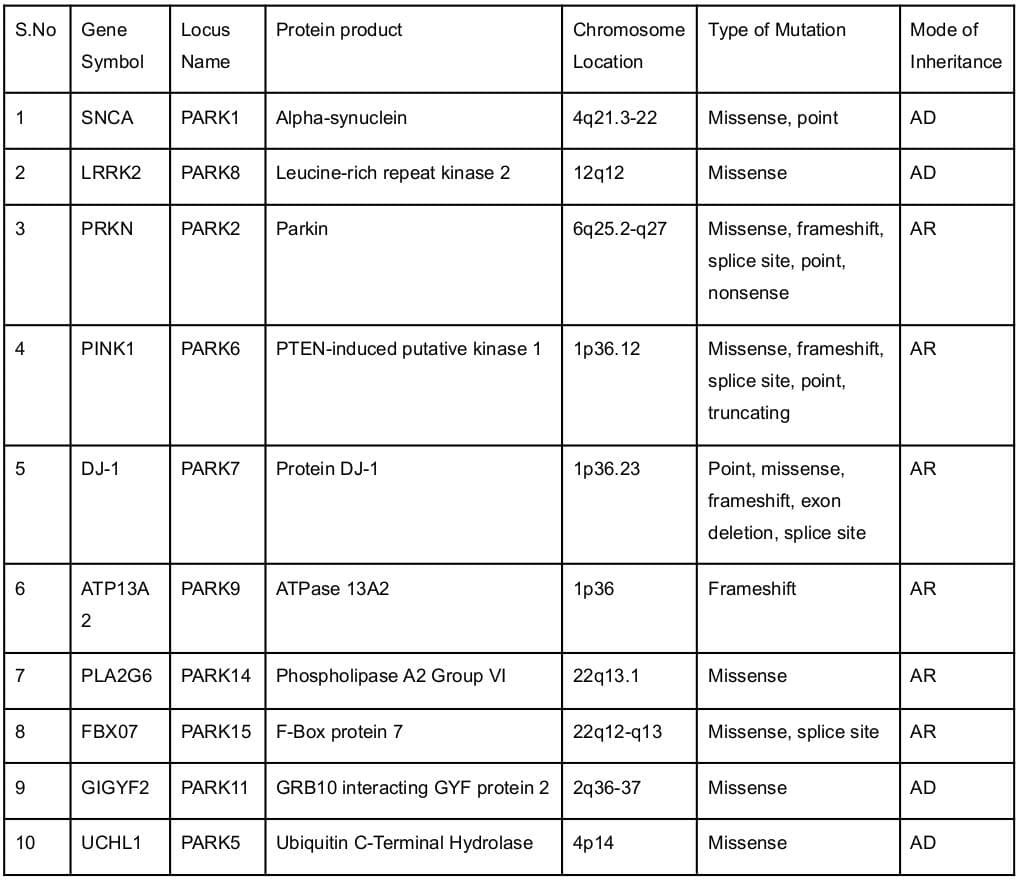
Significant strides have been made over the past decade in comprehending the genetic factors that contribute to Parkinson's disease (PD). An estimated 15 to 25 percent of PD cases exhibit a familial history of the disorder, and specific genetic mutations are accountable for around 30 percent of familial cases and 3 to 5 percent of sporadic cases lacking familial links. Mutations in certain genes, such as LRRK2, PARK7, PINK1, PRKN, or SNCA, are linked to familial PD cases. Additionally, variations in other genes like GBA and UCHL1 have been identified as modifiers of PD risk within specific families. However, the complete understanding of genetic alterations in relation to PD continues to be an area of ongoing research.
In familial cases of Parkinson's disease, the inheritance pattern varies based on the implicated gene. If the LRRK2 or SNCA gene is involved, the disorder adheres to an autosomal dominant pattern, where a single altered gene copy in each cell suffices to trigger the disorder. Thus, in most cases, an affected individual has only one parent with the condition. When PD is linked to the PARK7, PINK1, or PRKN gene, it follows an autosomal recessive inheritance pattern, necessitating both gene copies in each cell to carry a variant for the condition to manifest. While the parents of someone with an autosomal recessive condition each carry a single copy of the altered gene, they typically do not exhibit any signs or symptoms of the disorder. In certain instances, genetic mutations may not directly cause PD but can heighten susceptibility to the disease, particularly when other factors are at play.
But to further understand genetics in relation to PD, it can be helpful to take a deeper look into some of the genes involved.
List of candidate genes and susceptibility genes involved in Parkinson's Disease

GBA
Mutations in the gene encoding the lysosomal enzyme beta-glucocerebrosidase (GBA) are associated with Gaucher's disease, a lysosomal storage disorder. A multicenter study led by the NIH, encompassing over 10,000 individuals with and without PD, demonstrated that people with PD were more than 5 times as likely to carry a GBA mutation compared to those without the disease. Mutation carriers also exhibited a higher likelihood of receiving an earlier PD diagnosis in their lives and having a family history of the disease. Similarly, research indicates that ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) can influence the risk of developing Parkinson's disease.
LRRK2
The protein produced by leucine-rich repeat kinase 2 (LRRK2) is inherently a protein kinase. Variations in this gene constitute the most prevalent genetic cause of autosomal dominant PD and are implicated in around 10 percent of hereditary PD cases and about 4 percent of cases lacking a familial history of the disease. Investigations show that a specific LRRK2 mutation, G2019S, accounts for up to 20 percent of PD cases in specific groups, such as the Ashkenazi Jewish population. LRRK2 gene mutations appear to impact both protein synthesis and the disposal of unwanted proteins through multiple mechanisms. As a kinase enzyme, this gene tags molecules within cells with phosphate groups, regulating protein enzymes' activity. NINDS-supported researchers at Johns Hopkins University found that LRRK2 mutations accelerate the gene's protein tagging of ribosomal proteins, essential components of cellular protein synthesis machinery. This acceleration can result in an excessive production of proteins, ultimately leading to cell death. LRRK2 gene mutations are also believed to hinder autophagy, the process by which cells break down nutrients, recycle cellular components, and eliminate unusable waste. Autophagy is critical for removing damaged organelles and abnormal proteins. Additionally, LRRK2 gene mutations can impede a specific type of autophagy known as chaperone-mediated autophagy, which involves escorting damaged proteins to lysosomes for breakdown. When this process is compromised, the mutations could lead to the accumulation of toxic alpha-synuclein aggregates within cells. Researchers are investigating the potential for overriding LRRK2 gene mutations by reactivating the chaperone-mediated disposal system.
PARK7
The PARK7 gene regulates the production of DJ-1 protein. Mutations in this gene result in a rare form of early-onset Parkinson's disease. Although the DJ-1 protein appears to serve several functions, none are fully comprehended. Over 25 PARK7 gene variants associated with PD have been identified. Some of these mutations lead to an abnormally small DJ-1 protein or alter the amino acids involved in protein synthesis. The resultant altered protein is unstable and malfunctions or is non-functional. Other variants delete a substantial segment of the PARK7 gene, preventing the production of functional DJ-1 protein. One of the potential roles of this protein is to protect cells, particularly brain cells, from oxidative stress caused by an accumulation of unstable molecules called free radicals, which can harm or kill cells. Dopamine-producing nerve cells are particularly susceptible to oxidative stress. Additionally, the DJ-1 protein might function as a chaperone molecule, assisting in the correct folding of newly synthesized proteins into their proper 3D configuration and helping to refold damaged proteins. Disruptions in this process can lead to the toxic buildup of misfolded or damaged proteins and eventual cell death.
PINK1
Over 70 mutations in the PINK1 gene have been identified as causative factors in PD. PINK1 mutations typically underlie early-onset Parkinson's disease, which occurs before age 50. The PINK1 gene encodes instructions for PTEN induced putative kinase 1, a protein that appears to safeguard mitochondria from dysfunction during periods of cellular stress, such as heightened energy demands. Many PINK1 gene mutations result in a loss of protein function, leading to the selective death of nerve cells characteristic of PD. However, much remains unclear about the specifics of this mechanism.
PRKN
More than 200 PRKN gene mutations have been identified as triggers for PD. These variations are associated with the juvenile form of Parkinson's disease, emerging before age 20, as well as some cases of the more common late-onset form. The PRKN gene, one of the largest human genes, produces parkin protein, which aids in breaking down and recycling surplus proteins by tagging them with ubiquitin molecules. Ubiquitin serves as a signal to transfer these proteins to specialized cellular structures called proteasomes, where they are degraded. This system functions as the cell's quality control by disposing of damaged, abnormal, and surplus proteins. Additionally, it regulates the availability of proteins involved in crucial cellular activities like cell division and growth timing. Specific PRKN gene mutations lead to a parkin protein that is abnormally small and non-functional, rapidly degrading within cells. Other mutations introduce deletions, insertions, or changes in DNA building blocks (nucleotides), causing disruption. PRKN gene variations associated with Parkinson's often result in a loss of parkin activity. The full impact of PRKN gene mutations on Parkinson's disease remains a topic of ongoing exploration. It's believed that these mutations disrupt the ubiquitin-proteasome system, causing the buildup of unwanted proteins and impairing normal cellular activities, including dopamine production. Additionally, mitochondrial dysfunction in dopamine-producing nerve cells could contribute to the development of Parkinson's symptoms.
SNCA
Synuclein Alpha (SNCA) produces alpha-synuclein protein, which aggregates into Lewy bodies in the brains of PD patients. Mutations in SNCA are tied to early-onset PD, although more than a dozen SNCA mutations are linked to PD, they are relatively rare causes. These mutations can cause alpha-synuclein to misfold or lead to excess gene copies, resulting in elevated protein production. The abnormal and misfolded buildup of alpha-synuclein is a hallmark of PD, contributing to the formation of Lewy bodies within brain nerve cells. In a healthy brain, alpha-synuclein resides in specialized nerve cell structures called presynaptic terminals, which release neurotransmitters for neuron communication, crucial for normal brain function. While most PD cases aren't directly connected to alpha-synuclein mutations, abnormal and misfolded alpha-synuclein is present in nearly all PD cases, forming Lewy bodies in nerve cells, especially in the substantia nigra and other brain regions. The role of Lewy bodies in neuron death remains uncertain; they could either contribute to it or represent a protective response against the toxicity of alpha-synuclein aggregates. Researchers are intently studying alpha-synuclein's normal and abnormal functions and its interaction with genetic mutations linked to PD.

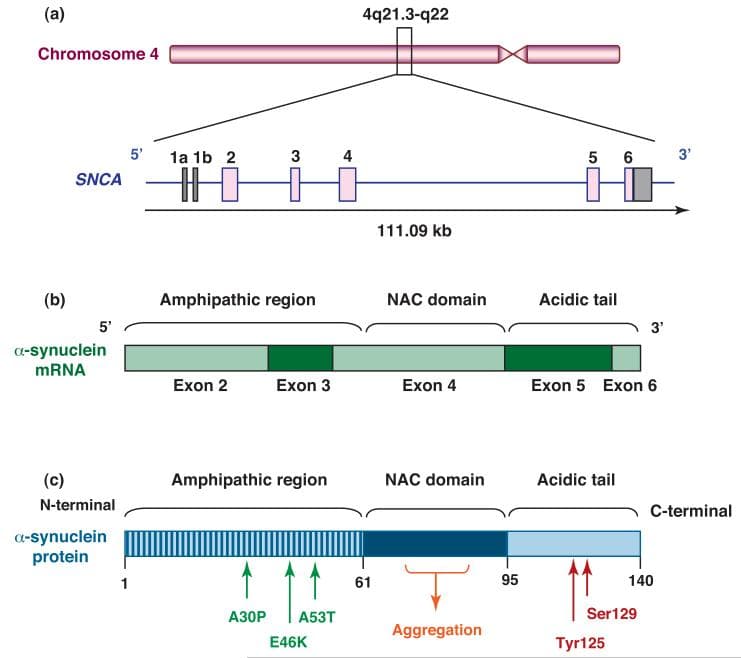
Schematic representation of human α-synuclein depicting: (a) SNCA gene structure, (b) mRNA, and (c) protein domain structure. Alpha-synuclein is a 140 amino acid protein and its sequence can be divided into three regions with distinct structural characteristics. The highly conserved N-terminal domain encodes for a series of imperfect 11 amino acid repeats with a consensus motif of KTKEGV reminiscent of the lipid-binding domain of apolipoproteins, which in certain conditions forms amphipathic helices. The six missense mutations described below lie in the amphipathic region, suggesting an important function for this region of the protein. The central hydrophobic region is associated with an increased propensity of the protein to form fibrils. The acidic C-terminal tail contains mostly negatively charged residues and is largely unfolded. ([Source](https://pubmed.ncbi.nlm.nih.gov/20961626/))

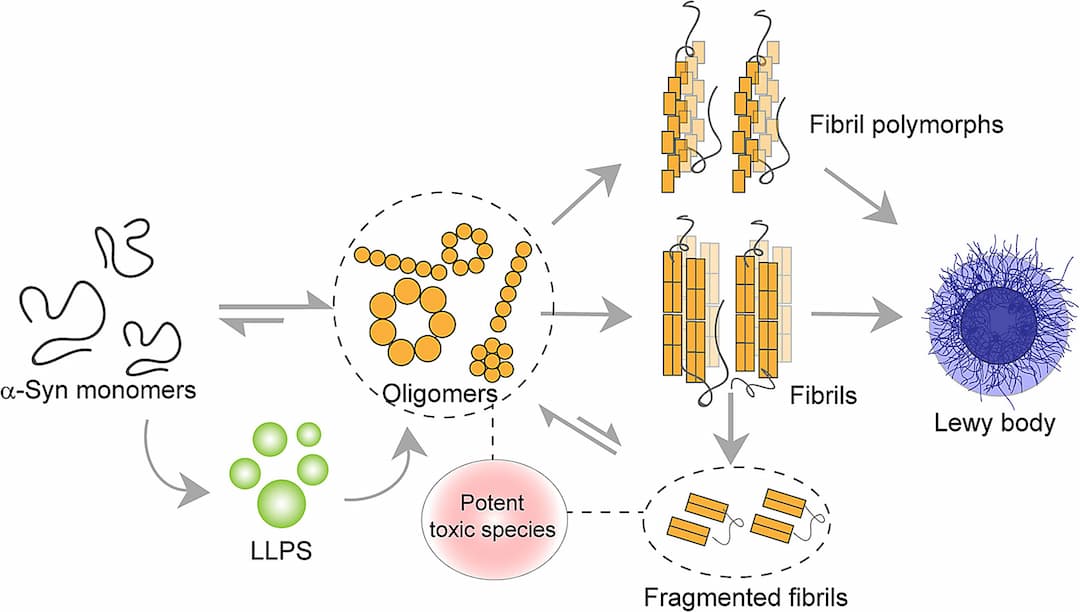
Alpha Synuclein aggregation leads to formation of lewy bodies. ([Source](https://www.sciencedirect.com/science/article/abs/pii/S0301462221002192))
Ongoing Research
There is a significant amount of ongoing research being conducted in relation to genetics and Parkinson's Disease, especially because so much is still unclear about the disease. According to the National Institute of Neurological Disorders and Stroke (NINDS), part of the National Institutes of Health (NIH), multiple NIH projects helped build an infrastructure for PD genetics research. The Human Genome Project and the International HapMap Project laid the groundwork for this research, producing tools to help researchers find genetic contributions to common diseases. With the support of these tools, scientists conducted the Parkinson's Disease Genome Wide Association Study (PD-GWAS). This collaborative initiative, funded by both the NINDS and the National Institute on Aging (NIA), aims to uncover genetic risk factors for Parkinson's disease from diverse populations worldwide.
The PD-GWAS dataset includes information from approximately 14,000 individuals diagnosed with PD and over 95,000 individuals without PD. Through a comparison of these two groups, researchers can identify specific regions, or loci, in the human genome where genes responsible for causing or increasing the risk of PD are likely to be found. These genetic loci can be likened to zip codes, providing valuable insights into the general neighborhood of relevant genes. Based on a combined analysis of PD-GWAS data and other sources, scientists have identified 28 loci believed to be independently associated with PD risk and many additional loci have been tentatively linked to the disorder.
In addition, recent advancements in next-generation genetic technologies have propelled discoveries in understanding the genetic factors contributing to Parkinson's disease (PD) risk. High-content genotyping initially yielded fruitful results, identifying common variants in the human genome. Currently, the focus is on next-generation sequencing, enabling rapid sequencing of specific genomic loci and significantly reducing the time and costs involved in pinpointing PD-related genes.
Another groundbreaking development in genetic sequencing is the NeuroX DNA chip, jointly developed by NINDS and NIA. This chip can identify genetic variants associated with the risk of late-onset neurodegenerative diseases, including PD. Despite these breakthroughs, more research is necessary to comprehend the roles of PD-related genes and their cellular processes in disease onset and progression.
Of particular interest are the PRKN and PINK1 mutations and their association with mitochondrial dysfunction, a crucial aspect to investigate in understanding PD. The interplay between PINK1 and parkin, both linked to mitochondrial processes, is being explored, with the hope of stimulating the PINK1/parkin pathway to induce mitophagy, which may lead to potential treatments for PD and mitochondrial diseases.
The impact of environmental factors, such as vitamin D levels, on PD risk is also being studied. Population-based research has indicated that higher vitamin D levels might offer protective effects against PD. However, genetic factors could influence how vitamin D affects PD risk, necessitating further investigation into the pharmacogenetics (the study of how an individual's genes affect responses to medications)of vitamin D to ascertain its potential benefits in PD prevention. Researchers are examining a large dataset to confirm the relationship between low vitamin D levels and PD risk while investigating potential genetic modifiers that may influence this association.
Deep brain stimulation (DBS), a medical intervention introduced in 1997 to alleviate Parkinson's tremor, received approval in 2002 for treating advanced Parkinson's symptoms. In 2016, it was further approved to address early-stage Parkinson's in individuals whose motor symptoms are effectively managed with medication. DBS involves implanting electrodes in specific brain regions that are responsible for motor control. These electrodes emit electrical impulses, modulating abnormal brain activity and mitigating Parkinson's symptoms. As research advances, DBS is emerging as a potential gateway for cutting-edge therapies like stem cell therapy and gene therapy in Parkinson's disease.
Combining gene therapy with DBS holds particular promise in addressing dopamine deficiencies in the brain.
Some studies have explored the possibility of using gene therapy to replenish dopamine levels alongside DBS, aiming for a more comprehensive and effective treatment approach. By introducing therapeutic genes into the brain, gene therapy can enhance dopamine production, potentially offering long-term benefits for Parkinson's patients.

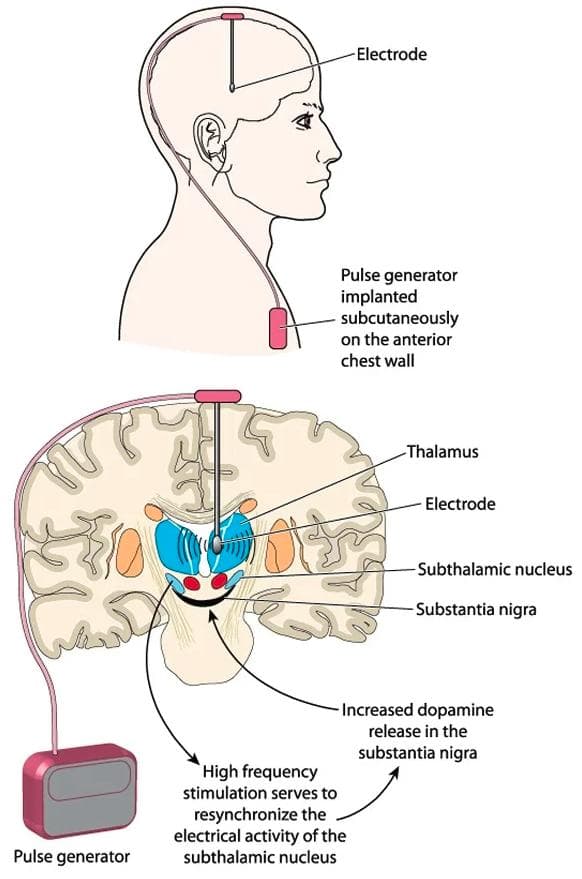
Shows further details of DBS. ([Source](https://www.news-medical.net/health/What-does-deep-brain-stimulation-involve.aspx))
Stem cell therapy:
Furthermore, researchers are increasingly optimistic about the potential of stem cell therapy and gene therapy as standalone treatments for Parkinson's disease. Stem cell therapy involves transplanting specialized cells, such as dopamine-producing neurons, into the brain to replace damaged or lost neurons. This approach aims to restore dopamine levels and improve motor function in Parkinson's patients. Gene therapy, on the other hand, focuses on altering the genetic makeup of cells to correct underlying genetic defects responsible for PD. By introducing functional genes or regulating gene expression, gene therapy aims to address the root causes of the condition, potentially leading to disease modification and improved symptom management.
As research progresses and technology advances, these innovative therapies may offer more targeted and transformative approaches to tackle the complexities of Parkinson's disease, ultimately improving the quality of life for those affected by this neurodegenerative disorder. However, continued research and clinical trials are essential to fully comprehend the safety, efficacy, and long-term effects of these novel treatments.
Future Direction
In the realm of genetics and Parkinson's disease research, promising future directions emerge. These include comprehensive GWAS to unearth new genetic variants, employing gene editing techniques to assess the impact of specific mutations on disease progression, and investigating gene-environment interactions. Analyzing gene expression profiles and exploring advanced imaging for potential biomarkers offer further insights. Personalized medicine and gene therapy may pave the way for targeted therapeutic interventions. By venturing into these paths, a deeper understanding of Parkinson's genetics can be achieved, and improved diagnostic and treatment strategies may be developed.
Conclusion
In recent times, there has been a remarkable leap forward in PD exploration. Pioneering researchers are diligently unraveling the enigmas of Parkinson's, and realistic ambitions now include treatments to restore lost function, impede the advancement of the ailment, and preempt its occurrence. Numerous promising therapies have been developed and are presently undergoing tests on both animals and humans. As scientists continue delving into the fundamental biology of the disease and the intricate interplay between genetic and environmental factors, they will uncover fresh biomarkers and further enhance therapies for alleviating PD symptoms, potentially leading to the ultimate halting, reversal, or prevention of the disease altogether.


Impact Statement

I’m Isabella Zarzar, a high school junior from the Bay Area who enjoys learning about biology and other scientific subjects. I decided to dive into Parkinson’s disease as it is a widespread neurological disorder that affects millions of individuals globally. Throughout this program, I learned a lot about genetics and human biology as well as how to conduct research thoroughly while utilizing various tools available on the internet. My experience with this project deepened my overall understanding of genetics and disorders that can be influenced by human genes. Additionally, I believe it enabled me to further improve my critical thinking and research skills in the future.
Student Reflection
References
National Institute on Aging. "Parkinson's Disease." National Institute on Aging, 2022, www.nia.nih.gov/health/parkinsons-disease.
"Parkinson's Disease: MedlinePlus Genetics." Medlineplus.gov, https://medlineplus.gov/genetics/condition/parkinsons-disease/.
"PARK7 Gene: MedlinePlus Genetics." Medlineplus.gov, https://medlineplus.gov/genetics/gene/park7/.
"PINK1 Gene: MedlinePlus Genetics." Medlineplus.gov, https://medlineplus.gov/genetics/gene/pink1/.
"PRKN Gene: MedlinePlus Genetics." Medlineplus.gov, https://medlineplus.gov/genetics/gene/prkn/.
"LRRK2 Gene: MedlinePlus Genetics." Medlineplus.gov, https://medlineplus.gov/genetics/gene/lrrk2/.
"SNCA Gene: MedlinePlus Genetics." Medlineplus.gov, https://medlineplus.gov/genetics/gene/snca/.
Kett, L. R., and W. T. Dauer. "Leucine-Rich Repeat Kinase 2 for Beginners: Six Key Questions." Cold Spring Harbor Perspectives in Medicine, vol. 2, no. 3, 3 Jan. 2012, pp. a009407–a009407, https://doi.org/10.1101/cshperspect.a009407.
"Parkinson's Disease: Challenges, Progress, and Promise | National Institute of Neurological Disorders and Stroke." Www.ninds.nih.gov, www.ninds.nih.gov/current-research/focus-disorders/focus-parkinsons-disease-research/parkinsons-disease-challenges-progress-and-promise.
Tran, Jenne, et al. "Genetic Predispositions of Parkinson's Disease Revealed in Patient-Derived Brain Cells." Npj Parkinson's Disease, vol. 6, no. 1, 24 Apr. 2020, pp. 1–18, www.nature.com/articles/s41531-020-0110-8, https://doi.org/10.1038/s41531-020-0110-8.
Gadhe, Laxmikant, et al. "Intermediates of α-Synuclein Aggregation: Implications in Parkinson's Disease Pathogenesis." Biophysical Chemistry, vol. 281, Feb. 2022, p. 106736, https://doi.org/10.1016/j.bpc.2021.106736.
Masato, Anna, et al. "Impaired Dopamine Metabolism in Parkinson's Disease Pathogenesis." Molecular Neurodegeneration, vol. 14, no. 1, 20 Aug. 2019, https://doi.org/10.1186/s13024-019-0332-6.
Xu, Shengli, and Piu Chan. "Interaction between Neuromelanin and Alpha-Synuclein in Parkinson's Disease." Biomolecules, vol. 5, no. 2, 5 June 2015, pp. 1122–1142, https://doi.org/10.3390/biom5021122.
By: Isabella Zarzar
The opinions expressed here are the views of the writer and do not necessarily reflect the views and opinions of Elio Academy.