Cystic Fibrosis
Understanding the Function and Dysfunctions of the CFTR Gene

Background
Cystic Fibrosis is a genetic disorder that is caused by a mutation in the Cystic fibrosis transmembrane conductance regulator ( CFTR ) gene. Cystic Fibrosis is an autosomal recessive disorder; therefore in order for a patient to have CF, the patient has to have acquired at least one mutated CFTR gene from each parent whether homozygous or heterozygous. The CFTR protein regulates several types of cells, such as the cells that produce sweat, mucus and digestive agents. Once the CFTR gene is mutated and dysfunctional, specific treatments and drugs are symptomatic. The idea of these treatments were introduced to me in school, and I grew to be more interested in theimpact of a gene mutation. This makes me curious to learn how professionals are able to consider each dysregulated part of the body to perform medical procedures that allow synthetic nature to regulate the bodies of CF patients.

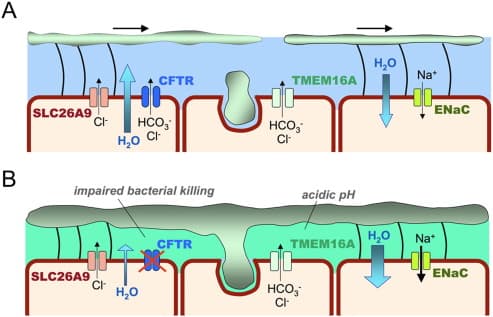
Molecular Physiology
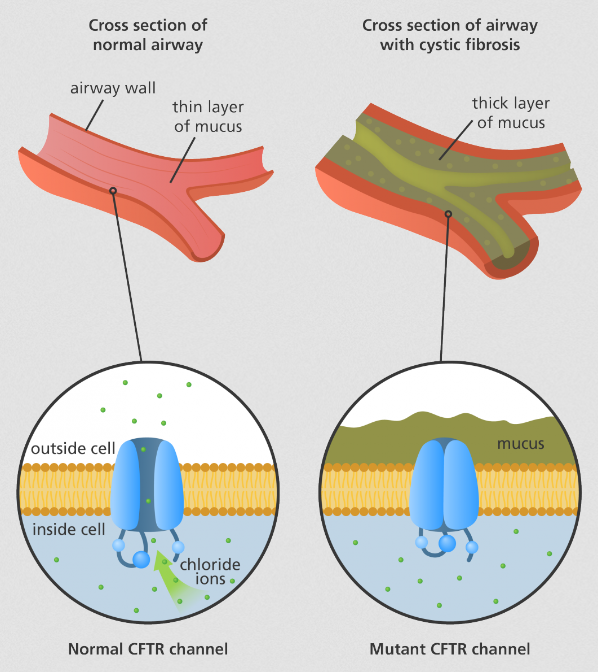
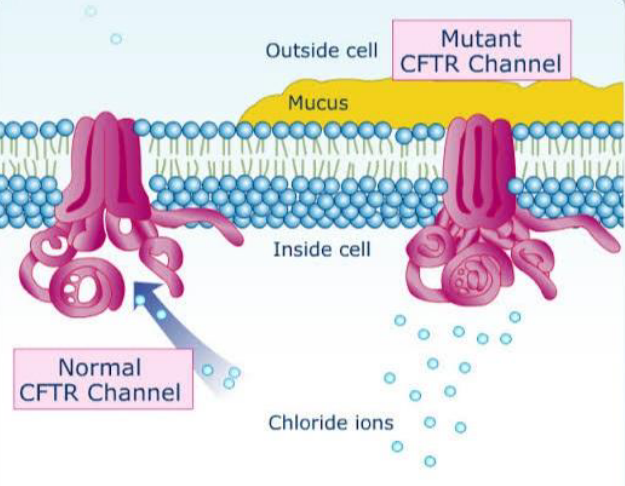
The CFTR protein is a channel that transports negatively charged chloride ionsthrough the cell membrane for internal and external access. In Cystic Fibrosis, the CFTR protein will be synthesized from a mutated CFTR gene and will be unable to perform regular functions. The mutation commonly seen in CF patients is the F508del mutation , resulting in the deletion of phenylalanine of the CFTR gene. The F508del deletion leads to the loss of 3 nucleotides that code for phenylalanine in the amino acid position 508. In a mutant CFTR protein, the protein will be defective in transporting negatively charged chloride ions in and out of the epithelial cells. The deletion of phenaylalanine in the CFTR allows the CFTR gene to be processed in an impaired post translational modification and therefore the channel protein leads to improper folding; the misfolded protein would be transported to the endoplasmic reticulum to degrade and not function for the body cells. When this occurs, sweat glands will be unable to absorb chloride ions back into the body, pancreatic juices will bedysfunctional in secretion and mucus will grow to be more viscous. Multiple treatments are being used currently to treat CF patients, ranging from physical therapy to drugs that secrete digestive enzymes to prevent malnutrition. Without proper activation of certain proteins such as the CFTR protein, parts of the body cannot be properly regulated and therefore leads to Cystic Fibrosis.

Considering the CFTR protein is commonly found, it can be located channeling chloride ions through cells in the digestive system, reproductive system and respiratory system. As discussed in class, protein function is heavily determined by protein structure and the synthesis of a mutated CFTR protein can affect how it functions as a channel protein. A regular CFTR protein is a multiplex protein with 3 domains. There are 2 nucleotide binding domains attached to each other with a transmembrane domain at the top, creating a barrel-like structure to allow chloride ions to pass through with ease. The transmembrane domain is a regulator and has to be phosphorylated in order to be activated and allow chloride ions to pass through the channel. If the F508del mutation is in the CFTR gene, the CFTR protein will go through animproper post translational modification due to an abnormal translation from the mRNA, the structure of the protein will be dysfunctional and therefore sent to be degraded.

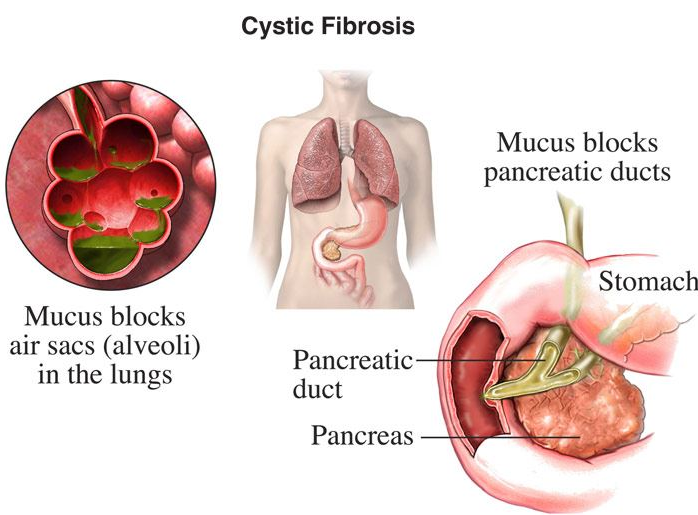
Disease Pathology
Most CF patients are diagnosed from a young age, considering the significant impact the buildup of mucus can have without regulation. This buildup of thick mucus can affect daily life for CF patients such as difficulty in breathing, malnutrition and sometimes infertility (in men).Thickened mucus would block airways in the lungs , where it would regularly be thinned out with the water molecules in the cilia. And mucus would also block the pancreas from secreting digestive enzymes to absorb and breakdown food in the intestines, making it difficult for patients to digest food properly.

Therapy
There are treatments and medication that are able to perform bodily functions to replace areas of the body affected by mucus , such as supplements that act as digestive enzymes that the pancreas secretes and medicines to make the mucus in the lungs less viscous. Although current treatments are effective enough for CF patients to live longer lives, medical professionals can also potentially look to more research for gene therapies that directly affect the mutated CFTR gene. Despite the current medication and treatments used to relieve CF symptoms, modulator therapies for Patients with the F508del mutation have been used in recent years to improve daily life. Therapies going by "Trikafta", "Orkambi' and "Symdeko" to name a few.
Although three therapies tackle the same genetic mutation, each therapy is individually selected for each patient as different correctors are used in every therapy. Most correctors used for CF are mixed in a combination in order to produce the best possible output in modulating a patient's genetic makeup. Elexacaftor/tezacaftor/ivacaftor , lumacaftor and ivacaftor are correctors used in the brand names of therapies listed. These correctors improve the function of the CFTR protein by improving the chloride channels or facilitating an increase of mature CFTR proteins.


By: Jaein Kho
The opinions expressed here are the views of the writer and do not necessarily reflect the views and opinions of Elio Academy.